用新柱子做了阿苯达唑的样品,然后走空白和溶剂空白,发现阿苯达唑的峰很高。请问大家,做阿苯达唑是不是容易在柱子上残留?还是其他原因?如果是柱子残留,什么柱子不易残留?
做PVC中的苯酚峰形异常,峰中间分叉,作标准物质也是,可能的原因有哪些?
大家好,最近使用HP-5检测邻苯二酚和对苯二酚,结果用乙腈作溶剂出峰正常,换成水作溶剂就不出峰了,很郁闷啊。真心求助各位大侠指点指点。 进样口和检测器温度300,柱温100-200,10℃/min.
求助!正常做出的标准曲线 在计算采样样品的时候 各个组份浓度都很正常 但是以甲苯计未知峰浓度特别大 是什么原因导致的
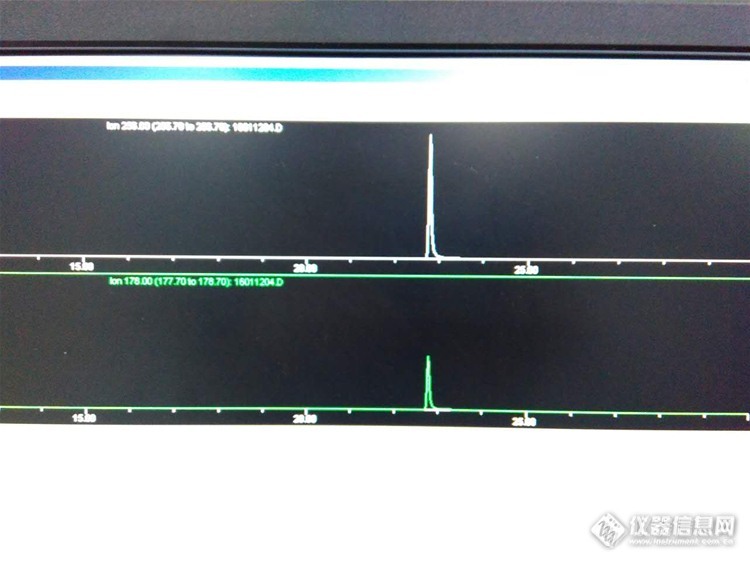
情况介绍设备:GC-MS 6890-5973(质谱配EI)柱子:DB-5MS检测项目:苯醚甲环唑(标准曲线)方法:GB/T 19648-2006 水果和蔬菜中500种农药及相关化学品残留的测定气相色谱-质谱法(下称19648)问题如下: 皮下来公司这三年该设备主担当项目为农药残留检测,极偶然的时候客串其他项目,所用的检测方法基本就两种:韩国食品公典(公司要求),GB/T19648-2006(CNAS要求)。最近领导要求追加检测种类,首先选择的就是苯醚甲环唑,皮下先确认了19648中的种类,发现该农药在其中,于是尝试用标准物质试验。 用5ppm进行scan,未发现明显峰形,于是用5ppm进行sim(323,235,265),结果总是有杂峰影响,尝试多遍仍然分不开,疑似有污染,用带内标的低浓度测试,结果内标峰形良好。然后搜寻其他m/z,发现有发现SNT 1975-2007 (质谱配CI)中有348,310,350,345,强行尝试。。。。。果然失败。。。。。 求助版友帮忙分析原因,或者有没有更好的分析方法。万分感激!!!!补充说明下,皮下晓得这个农药本就是有两个峰的,可是例如氯氰菊酯那样有四个峰的,我们的机器也可以分的很清晰,可是这个的两个峰是完全在一起了T.T要更准确的定性和定量还是要分开吧T.T 求助大家~谢谢~1、标准品5ppm无内标(323,325,265)http://ng1.17img.cn/bbsfiles/images/2016/01/201601140949_581821_2669685_3.jpg2、1ppm内标(265、178)http://ng1.17img.cn/bbsfiles/images/2016/01/201601140950_581822_2669685_3.jpg
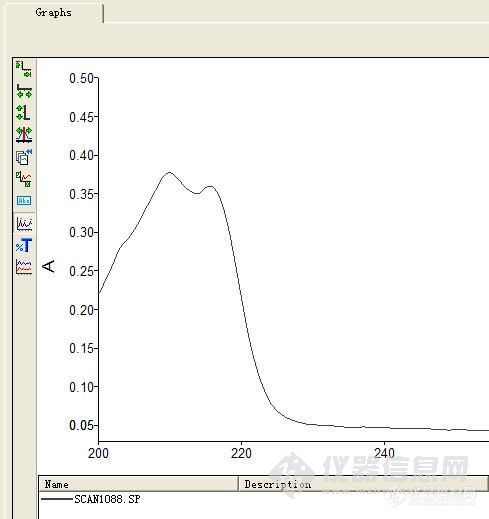
2004年买的一台PE Lambda 35 分光光度计,测定苯酚时,吸收峰从原来的270nm跑偏到210nm,打开机箱看了一下,啥也动不了!请问怎么回事?[em09512][img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912101604_189311_1604910_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912101606_189314_1604910_3.gif[/img]
为何我用SE-30做苯系物,苯的峰出的特别不好看,其他峰还挺好,对二甲苯和苯乙烯分的不是特别好,响应峰高特别小,怎么办呢?
我用的Agilent的7890GC,做的是甲醇中7种苯系物的含量测定,分别为苯、甲苯、乙苯、对二甲苯、间二甲苯、邻二甲苯、苯乙烯。柱子是用的瓦里安的52CB的毛细管柱,30m*0.32mm*0.45um,为了把对间二甲苯分开,设了个升温程序,50度保持4分钟,然后升温到80度保持至结束,载气流速2ml/min,分流比设的50。之前也试过别的条件,发现甲醇的溶剂峰非常大,苯的出峰总是在甲醇的溶剂峰的拖尾上,可能给定量带来较大误差,请问这个怎么解决?(换过手头的Waxetr柱,分对间二甲苯效果还不如这个柱,别的柱子更不行)
最近做五氯苯酚,不论是进标样还是样品,五氯苯酚乙酸酯都是出两个相连的峰,离子峰都是五氯苯酚乙酸酯,老司机指导指导是啥原因这个应该没同分异构吧= =
有没有谁苯酚用气相色谱做的,希望能说一下方法呀,自己用顶空做了,检测器是MS,发现找不到苯酚的峰,我们的柱子型号是Elite-5MS,
最近在做化妆品测定苯酚和丙烯酰胺的实验,按照标准的方法,苯酚采用C18柱,甲醇与水(60:40)等度洗脱,1ml/min,280nm;标准方法里,苯酚的保留时间的4分多,但是我做的在不到1分钟就已经出完峰了,感觉不太对。请教下,有没人做这方面的,给点建议,谢谢!
急急急,请教各位老师,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做苯系物,分流比为20时,各个物质出峰峰面积特别小,0.25ppm的检测不到峰,但是溶剂峰特别大,峰面积有一百三十多万。如果继续改分流比,会导致溶剂峰更大,会不会对衬管进样口有影响呢?
最近我们客户方面检出我们产品中有苯醚甲环唑,于是我们也增加了这个项目。但是我们根据相关系列上的设置进行检测时,他们的检测限能达到0.001左右,但是我们的一直在0.1左右。仪器设置和他们一模一样也达不到他们的检测限。找了几份做苯醚甲环唑的资料,按他们的方法做也是达不到0.001的检测限。直接进标准品的话,0.5ppm左右才能有比较好的峰形。我们用的是安捷伦的GC6890N/MSD5975C,EI源;仪器是正常的。有没有哪位高手给指点一下。有做这个项目的朋友帮忙发份仪器设置和前处理方面的资料,谢谢1217976244@qq.com
我在用HP-INNOWAX极性柱分析苯酚,苯酚的峰还是拖尾,而别人用同样的柱分析苯酚,峰非常漂亮,很尖锐的峰。这是为什么?谢谢。
各位大神,最近用顶空做苯,怎么都不出峰,顶空条件60度,恒温30分,色谱进样针和检测器都设的200度,柱温50度,各位帮我看下条件还需要怎么优化下呢,气相是安捷伦的7890
苯酚标准是甲醇中的苯酚。配标准时解析液是丙酮,要是按标准方法里面的条件来做的话,丙酮和甲醇是分得开,不过,苯酚的出峰时间会在30分钟左右。其实我们在没有必要把丙酮峰和甲醇峰分开的时候,我调了条件,苯酚在四分左右就可以出峰了,你们说我这样做是可行的吗?
求教丙酮做溶剂测苯酚,出峰的时间段内会有杂峰,具体怎么区分,并且峰面积特别少
依据木制品五氯苯酚的测试标准。 五氯苯酚乙酰化后, 用GC-ECD检测,DB-17的毛细管柱,升温程序是初始温度50摄氏度(1min),以15摄氏度/min程序升至300摄氏度。奇怪的是: 1)正己烷作为稀释剂,为空白溶液,竟然有三个色谱峰; 2) 标准物质(PCP)和内标物(三溴苯酚)同时乙酰化,内标物出峰,可是五氯苯酚乙酰物不出峰,试过DB-5的柱子,改变初始温度和升温程序都不出峰。急切需要帮助,万分感激!
[table=100%][tr][td]我用HPLC测对苯二酚的甲醇溶液,在2-3保留时间内出现两三个峰,而且是肩峰,没有分开。但在同条件下用对苯二酚水溶液做就只有2.8左右的一个峰,而且峰型非常好。对苯二酚纯度为98%,甲醇是色谱纯的,分析条件是流动相:甲醇:水=20:80,波长:290nm,流速1ml/min。求高人指点!![/td][/tr][/table]
看到国标GB/T 14677-1993空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量 甲苯、二甲苯、苯乙烯的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法中,做苯的标准曲线时要用二硫化碳做溶剂,我看到有人也用甲醇做溶剂,溶剂的不同会有影响吗? 另外在这个标准里面,不管是标样还是被测气体样品,都用采样管来处理然后通过加热解析进入色谱。我们这里装置简陋,可以在做标准曲线的时候,直接向气化室进配好的标准溶液,在做被测气体的时候,直接通过六通阀进气样吗,然后根据被测气体出峰的面积在标准曲线上找对应的含量吗? 希望了解的人回答,万分感谢!很感谢大家的回答,但还是没有我想要的答案,抛开国标不说,如果我用甲醇或者二硫化碳为溶剂配置了一定浓度的苯溶液,假设为1mg/mL,然后用微量注射器取1、2、3、4、……uL进样,这样苯的进样质量就是1、2、3、4、……ug,以进样质量为横坐标,峰面积为纵坐标作图。然后在后面测空气样品时(直接空气进样),可否将前面的图作为标准曲线用,通过测得的峰面积得到进样中苯的质量,然后根据进样体积得到苯的浓度?写的有点长,感谢大家耐心阅读并作出回答。
最近做阿苯达唑,有几个问题想请教下:1.取样量问题:精密称取适量(约相当于阿苯达唑20mg),这个取样量不是20mg,因为阿苯达唑药片里含有其他辅料,所以取样量应该比20mg多,对吗?2.试剂问题:用乙醇稀释至刻度,这个乙醇是无水乙醇吗?3.结果偏低,是什么影响的?
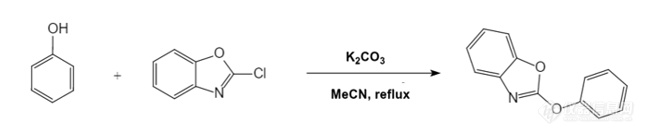
求助各位大佬,请问2-氯苯并噁唑与苯酚具体的反应步骤是什么?苯酚阴离子在进攻2-氯苯并噁唑时双键打开吗?还是氯离子直接被取代?[img=,646,135]https://ng1.17img.cn/bbsfiles/images/2023/04/202304101159588512_6104_5971175_3.png!w646x135.jpg[/img]
做水质中的苯系物,苯峰和甲醇峰分不开,怎么办?做了一个星期了,还是不行,调分流比和温度都分不开,怎么办?
有没有做苯甲酸,山梨酸的高手啊,想请教下,今天用GB5009.29的方法做苯甲酸和山梨酸,但是不管我怎么做标准品都只出来一个峰,我以为时间推迟了,加长时间到25分钟,用2ml\min的流速,但是也只有一个峰,应该是山梨酸的峰,上个月我还测过,是可以做的,难道是我的标液过期了吗,按理说它们作为防腐剂应该没有那么容易分解的吧?我的100ppm的标液是3月份配制的。 我以为是以前有什么问题 ,开始是用DAD检测器,后来换了台用UV检测器检测,但是结果也一样,就只有一个峰,明天想重新配制标液看看,但是不知道是不是标液问题,有没有人能帮我分析下可能有哪些问题会引起这样的现象?谢谢了
经常遇到氘代氯仿的7。26峰与样品苯环的峰重叠。无法分开。此时如何积分?还积不积7。26的峰。谢谢
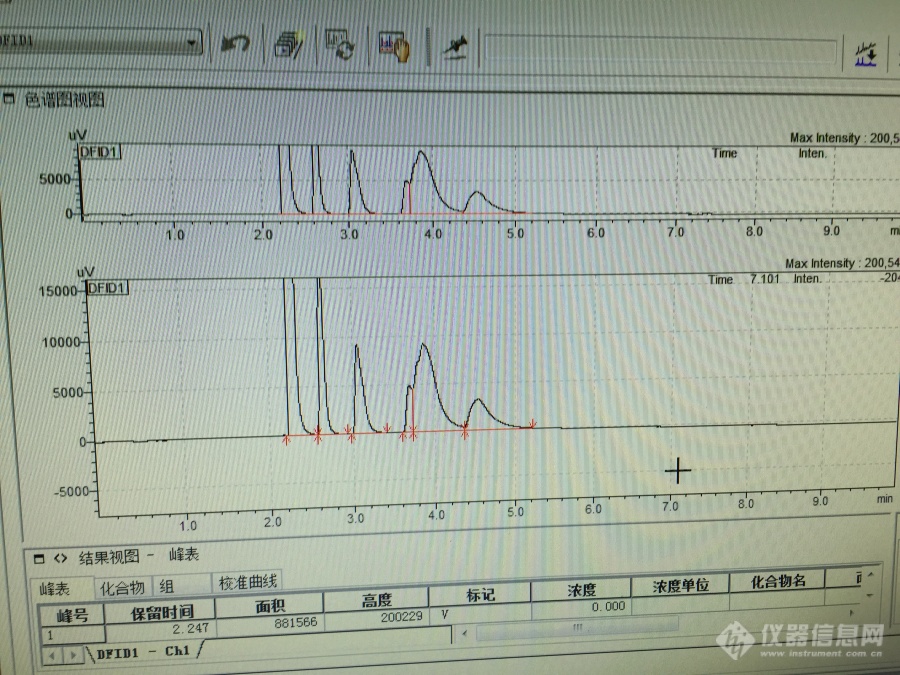
做水质苯系物6项时,为什么色谱峰这么奇怪,有些峰怎么分都分不开?请问什么问题?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907161315531158_6946_1815404_3.jpg!w690x517.jpg[/img]
求下面依达拉奉注射液中苯肼检测的专利和文献。??????谁可以下载的帮我下载一下吧,求求了[img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308141926478314_1702_5341064_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308141926479682_8480_5341064_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/08/202308141926480553_6273_5341064_3.png[/img]
请问有做邻苯的高手吗?特别是做玩具的,我用的是气质我刚做这个不久,请教一些问题。我是按国标做的,用索氏提取每次做的量有限,有别的更好的方法或标准吗?DINP和DIDP出来的是五指峰,我要怎么定量,手动积分总积不好导致标液曲线线性不好,网上有人说把他们两个的峰面积加起来,那如果是要很准确定量呢?还有就是怎么看响应值。对于这些问题一直不知怎么解决,感谢帮助的人。

发现做三苯时候苯峰被干扰的问题,特发一帖,与各位分享。昨天在做三苯样品时候发现空白二硫化碳中含有少量苯,如下图一:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091717_531833_2780210_3.png一开始以为是二硫化碳被污染,本人遂再开一瓶,进样后发现仍然有苯峰,如下图二:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531834_2780210_3.png为再次确定结果,正好手头GCMS空闲,上GCMS上走Scan以及SIM后发现没有苯的定量离子,所以我得出的结论为,二硫化碳未被污染,而是GC问题。立即动手,更换耗材,老化柱子,老化检测器。老化过程中发现基线有一些杂峰出来,让我更加坚信是GC端的问题。当我开开心心的再次走三苯样品的时候,悲剧出现了,苯峰没有被完全消除!如下图三:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531835_2780210_3.png今天接了一个要分将对/间二甲苯分开的单,所以拿出好久没用的DB-WAX的柱子,在另外一台GC上老化后直接上曲线走样,在做空白的时候惊讶的发现万恶的苯峰又来了,我判断问题仍然处在二硫化碳上。可是同一瓶二硫化碳走过MS,MS的灵敏度总不会低于FID吧?在思考的时候,样品走到1ppm浓度的三苯标液上,我发现苯峰分叉了!如下图四,图五:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531836_2780210_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531837_2780210_3.jpg证明空白中的那个被当做苯的杂质并不是苯,是一种跟苯有着相同保留时间的另一种物质!突然想起又一次其他实验室的过我们实验室来交流的时候有说过,正己烷有可能干扰到苯的测定。回想一下,早上更换装WAX柱子GC的洗液时正好看洗液是做有机氯农药时候用的正己烷!立即拿着刚测定有苯的空白进GCMS走了一个Scan,如下图六:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531838_2780210_3.png正己烷 57 43 的离子果然在!终于发现问题所在了!各位,要小心正己烷冒充苯啊!刚刚很多老师建议说极性柱子正己烷出峰应该在前面,这的确是我没想到的地方,找个空闲时间进一针正己烷试试,谢谢各位老师提醒!刚刚我用相同的条件进了一针正己烷,出峰时间在二硫化碳后面一点!看来还是我考虑得不周到!
苯醚甲环唑在hp-5 113-2018的方法出峰大概33min左右,但出现拖尾比较严重,定量比较困难?有哪位老师有解决办法,求指导







 我要推广仪器
我要推广仪器
 下载APP
下载APP