增效联磺片为磺胺类抗菌消炎药的新型复方制剂,每片含磺胺甲基异(口恶)唑200mg、磺胺嘧啶200 mg、甲氧苄氨嘧啶80 mg,各地方标准均有收载,对前两种成分以纸色谱法鉴别,而对甲氧苄氨嘧啶则另行鉴别。本文以薄层色谱法同时鉴别磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶3种成分,专一性强,斑点明显,操作简便,结果较为满意。1 仪器与试药 三用紫外线分析仪(上海顾村电光仪器厂),硅胶GF254薄层板(10cm×20 cm,自制);磺胺甲基异(口恶)唑、磺胺嘧啶和甲氧苄氨嘧啶对照品(中国药品生物制品检定所);增效联磺片(市售品);硅胶GF254(青岛海洋化工厂生产,化学纯);其它试剂均为分析纯。2 溶液的配制2.1 单一对照品溶液 分别精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液分别制成0.5 mg/mL磺胺甲基异(口恶)唑、0.5 mg/mL磺胺嘧啶、0.2 mg/mL甲氧苄氨嘧啶的单一对照品溶液。2.2 混合对照品溶液 精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液制成? mL含磺胺甲基异(口恶)唑0.5 mg、磺胺嘧啶0.5 mg和甲氧苄氨嘧啶0.2mg的混合对照品溶液。2.3 样品溶液的配制 取供试品细粉适量(约相当于磺胺甲基异(口恶)唑50mg),加50 %丙酮溶液100 mL,振摇使溶解,过滤,滤液作为供试品溶液。

测N-(甲氧甲基)-N-(三甲基硅甲基)苄胺我采用岛津[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],柱子为wax(聚乙二醇),所设参数SPL 290度,DFID 290度,柱温40度下保留1 min再以10度每分升温到240度,直接进样走到中间在某一峰后出现基线严重漂移,但基线可以回到零点,用甲醇稀释后基线漂移没有那么明显,而此处也显示有另外一个物质存在,想请教高手出现这种状况是什么原因?是物质沸点太高参数选择不对还是物质与柱子极性不匹配?N-(甲氧甲基)-N-(三甲基硅甲基)苄胺的沸点为76 º C (0.3 MMHG)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181274]N-(甲氧甲基)-N-(三甲基硅甲基)苄胺色谱图(甲醇溶解).doc[/url][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911051117_181361_1618994_3.jpg[/img]
请告诉2-甲基-4-甲氧基-二苯胺的液相色谱分析方法
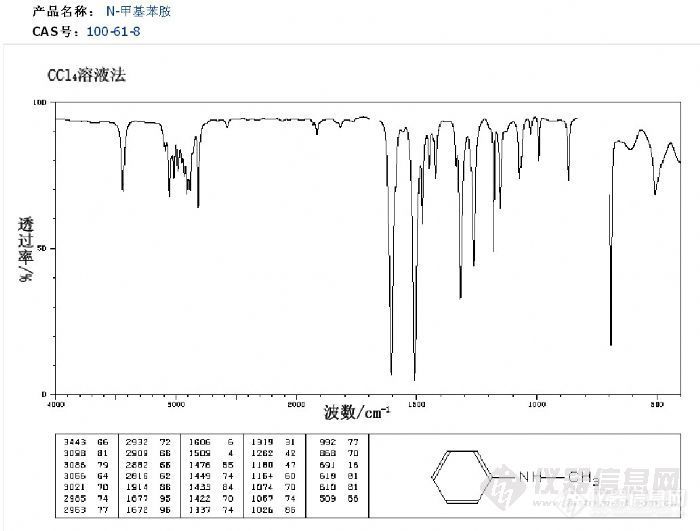
单位是做汽油检测的,最近要上新项目做汽油中N-甲基苯胺,甲缩醛2种添加剂,用的是中红外,初步查到的信息是N-甲基苯胺,的定性是在红外谱图中有检出甲基胺的吸收峰,则判断N-甲基苯胺检出。没接触过红外,不知道怎么看红外谱图中甲基胺的特征吸收峰。附上一张N-甲基苯胺的红外图。希望有懂的 给点建议http://ng1.17img.cn/bbsfiles/images/2012/03/201203221523_356642_2052186_3.jpg
如题,如果不能打色谱,那有没有其他的检测方法能检测出甲基羟胺和二甲基羟胺的含量。多谢各位大虾~~甲基羟胺和二甲基羟胺是甲氧胺生产过程的副产物。
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
各位大侠帮帮忙,从文献上看到如题衍生化试剂,但上面说取适量4-溴甲基-7-甲氧基香豆素和18-冠醚-6溶于N,N-二甲基甲酰胺10ml,可到底取多少量呢,各位有知道的吗,帮帮忙,具体这个衍生化试剂怎么配得呀?急候大家的回复!谢谢啦!
亚硝基二甲胺和二甲基亚硝胺是不是一种东西?
氮甲基苯胺、邻甲基苯胺、间甲基苯胺,对甲基苯胺在用红外光谱检测时,波长分别是多少
分析对甲基苯胺,间甲基苯胺,邻甲基苯胺,用什么毛细管色谱柱好?
哪位同道有 对氨基二甲基苯胺比色法 GB 18056 –2000 附录A 用来检测甲硫醇的,急需!!!
1701柱,一个是DB,一个是VF1701MS,尺寸是一样的,30*0.25*0.25,可是DB1701上甲基异柳磷与水胺硫磷能分开,相差1.0min,而VF1701上只相差0.1min,两者重合,大家遇见过这种情况吗?
我用的是250mm的C18柱,紫外检测器,检测波长243和280nm,流动相:甲醇和10%乙酸溶液,梯度洗脱,柱温40℃,流速0.8mL/min,进样量:20μ,测藜芦醛和N-甲基甲酰苯胺,这俩峰咋分不开,是哪里的问题啊,恳请各位大佬指教
分析对甲基苯胺,间甲基苯胺,邻甲基苯胺,用什么毛细管柱色谱柱好?
最近要测n甲基甲酰胺标准品但是找了一些标准品厂家都没这个标,要证书的,所以也不能用溶剂代替。有买过的或者有货号的请提供一下给我
各位老师好:我目前做N,N-二甲基乙二胺溶剂的残留时出现奇怪的现象,配制5000ppm的N,N-二甲基乙二胺标准溶液,有两个色谱峰,前面的小后面的大;但是我用N,N-二甲基乙二胺的储备溶液(由该溶液配制的5000ppm的N,N-二甲基乙二胺标准溶液)进样时,却变成前面的大后面的小!请老师指导!谢谢
各位用3355四甲基联苯胺测余氯的同行:有几个问题欢迎大家讨论:1.你们用的3355四甲基联苯胺溶液是按GB5750.11-2006标准配置的吗?配制时有将其中的0.1mol/lHCL浓度加大的没有?2.你们的水样PH一般是多少?有需要调整的吗?大概PH多少时需要调整?3.调整时是先加1+4HCL再加水样调节好PH后再加入3355四甲基联苯胺溶液,还是将1+4HCL和3355四甲基联苯胺两种一起加入比色管再加入水样比色的?或是直接使用已加大HCL浓度配制的3355四甲基联苯胺溶液再加入水样检测?4.有做过用3355四甲基联苯胺比色法和HACH便携式或在线监测(DPD试剂)检测法对照试验的吗?两种方法测定结果怎样?谢谢各位发表高见!
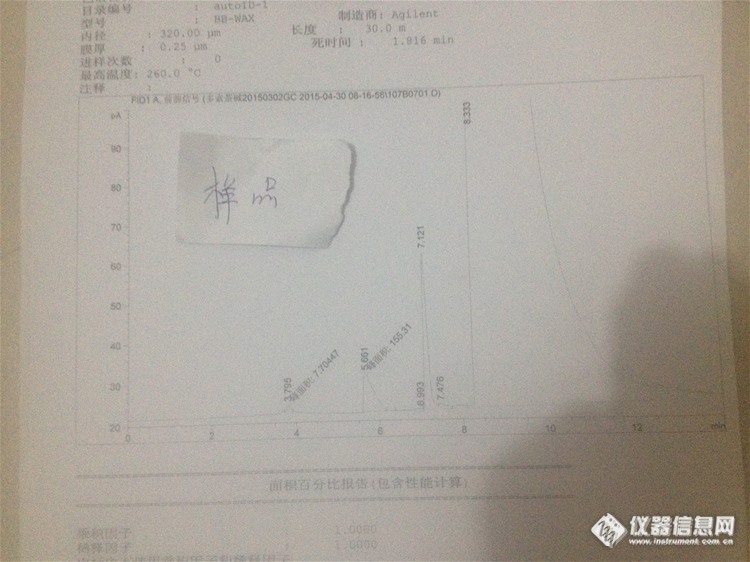
大家好,我最近用气相色谱做乙醇和N,N-二甲基甲酰胺残留溶剂分析的时候遇到了点问题,想向大家请教一下!问题如下: 图一是乙醇的定位峰,主要是乙醇和作为溶剂使用的二甲基亚砜 图中乙醇峰保留时间为3.357http://simg.instrument.com.cn/bbs/images/default/em09504.gifhttp://ng1.17img.cn/bbsfiles/images/2017/10/2015052011030483_01_2991692_3.jpg 图二是N,N-二甲基甲酰胺的定位峰,主要是N,N-二甲基甲酰胺和作为溶剂使用的二甲基亚砜 图中二甲基甲酰胺保留时间为6.031 http://ng1.17img.cn/bbsfiles/images/2017/10/2015052011033035_01_2991692_3.jpg 图三是乙醇和N,N-二甲基甲酰胺的对照(乙醇浓度大概500ug/ml,二甲基甲酰胺浓度大约为88ug/ml) 图中可以判断乙醇峰保留时间3.539 这里出现了分歧,可以判断甲酰胺的峰有 一个是保留时间为5.617峰面积176.7 另一个保留时间为7.381 峰面积 88.6 还有一个是7.079 峰面积383.5 。http://ng1.17img.cn/bbsfiles/images/2017/10/2015052011034766_01_2991692_3.jpg 图四为样品,处理方式为样品加二甲基亚砜 样品中保留时间5.655 峰面积155.7 保留时间7.115 峰面积257.2 保留时间7.437 峰面积35.9 http://ng1.17img.cn/bbsfiles/images/2017/10/2015052011041558_01_2991692_3.jpg 图五为样品+少量的二甲基甲酰胺,二甲基亚砜为溶剂。保留时间5.633,峰面积327.4 保留时间7.150 峰面积296.4 保留时间7.361 峰面积217.7 http://ng1.17img.cn/bbsfiles/images/2015/05/201505201104_546778_2991692_3.jpg那么,问题来了,怎样判断对照里面哪个峰是N,N-二甲基甲酰胺? 请大家帮忙分析一下!谢谢!
EP里面做N,N-二甲基苯胺的方法A里规定的毛细管色谱柱填料是:[color=#333333]交联聚甲基苯基硅氧烷(cross-linked polymethylphenylsiloxane R)。网上找了半天没找到具体型号,不知道大家有没有人知道这是什么型号的色谱柱?[/color]
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
1. 在用亚甲蓝测水中硫化物的时候,需要配置0.2%的对氨基二甲基苯胺溶液,我查了一下,他的别名也可以叫叫[font=宋体, Arial, Helvetica, sans-serif][size=12px]N,N-二甲基对苯二胺二盐酸盐,应该没买错吧。[/size][/font]2. 他的储存条件不是-20℃吗?这样它里面会不会含有很高的水分?那我称取2 g的 时候,需不需要干燥,还是直接就称取2 g 就好了。3. 还有就是配置好0.2%的对氨基二甲基苯胺溶液后,怎么储存呢?室温?冷藏?冷冻?如有回复,万分感谢!![img=,690,449]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239193272_1018_5383916_3.png!w690x449.jpg[/img][img=,690,299]https://ng1.17img.cn/bbsfiles/images/2021/10/202110221239240158_2694_5383916_3.png!w690x299.jpg[/img]
最近用hp-5的色谱柱分析N,N-二甲基乙酰胺和其他的一些残留溶剂,溶剂采用二甲亚砜。N,N-二甲基乙酰胺不出峰,不知道是不是因为二甲亚砜和N,N-二甲基乙酰胺重合了的缘故。对了我的色谱条件是顶空程序升温,40度保持10分钟,10度每分钟升温至210度。二甲亚砜出峰时间在11分钟,较大的拖尾溶剂峰,大家帮忙看看是什么情况?
如题,鱼类的磺胺类药物,抗生素,甲醛,甲基睾酮,呋喃唑酮的含量一般是多少啊,哪位高手有做过类似的能否告知一下,我做出来的磺胺类磺胺嘧啶、磺胺甲基嘧啶、磺胺5甲氧嘧啶、磺胺二甲基嘧啶、磺胺甲基嘧啶的总量有100多ug/kg,好像很高,找不到对比啊,哪位大侠帮帮吗,救命啊
30m×0.32mm,1.0μm规格的5%苯基-95%甲基聚硅氧烷色谱柱,可分离甲苯和二甲基甲酰胺吗?
大家测余氯的时候四甲基联苯胺都是怎么配置的?按照国标吗?盐酸配置0.1摩尔每升100毫升怎么配啊?
有没有做过N-亚硝基胺的?我用N-二甲基亚硝基胺标准物质进样,但是根本观察不到信号,TIC和SIM都没有信号,这是什么原因呢?
依照GB5009.26-2023第一法,氮二甲基亚硝胺需要蒸馏,如果买类似凯氏定氮仪的蒸馏装置,价格要10万左右,太贵了,自己搭建的话,也不切实际,请问下大家用的什么样的蒸馏装置,从哪里可以买到成套的搭建的玻璃装置,这样性价比更高一些?谢谢
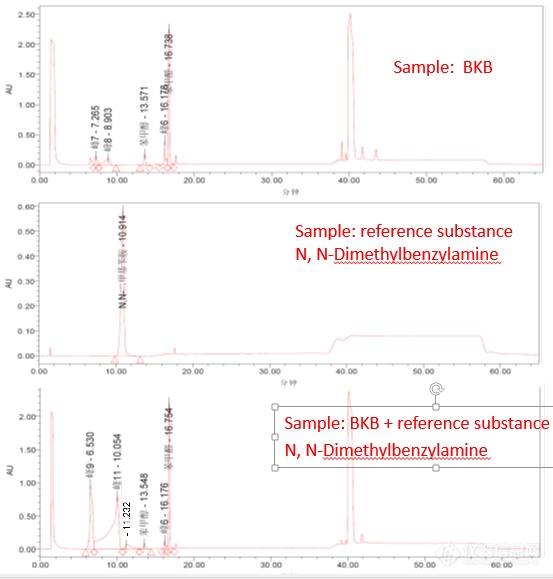
[color=#444444]最近在测苯扎溴胺(BKB)中苄基二甲基胺的残留,在测试条件下标样苄基二甲胺能够正常出峰,而进BKB样品时,苄基二甲基胺却不出峰,在BKB样品中加入苄基二甲基胺标样,却出现了两个奇怪的峰见色谱图,不知这是什么情况?[/color][color=#444444]色 谱 柱:端基封闭的C18(150×4.6mm,5μm)。[/color][color=#444444]柱 温:30℃[/color][color=#444444]流动相A:磷酸盐缓冲液(取己烷磺酸钠1.09g、磷酸二氢钠6.9g,溶于适量水中,用磷酸调节pH至3.5,用水稀释至1000ml,摇匀,即得。[/color][color=#444444]流动相B:甲醇[/color][color=#444444]时间(分) 流动相 A(V/V) 流动相 B(V/V)[/color][color=#444444]0-10 80 20[/color][color=#444444]10-14 80-50 20-50[/color][color=#444444]14-35 50 50[/color][color=#444444]35-36 50-20 50-80[/color][color=#444444]36-55 20 80[/color][color=#444444]流 量:1.0ml/min[/color][color=#444444]检 测 器:二极管阵列检测器(DAD)[/color][color=#444444][img=,553,579]https://ng1.17img.cn/bbsfiles/images/2019/06/201906031402122584_6731_1823055_3.png!w553x579.jpg[/img][/color]
公司要测PU皮中的二甲基甲酰胺残留,送外ITS几次,检测数值一般是30-60PPM左右,检测公司用的是顶空进样法来测试。我们没有顶空设备,只有GCMS,所以我采用甲醇萃取,进样萃取液的方法来测试,基本在150-250ppm左右。问题一:萃取法比顶空法数值大这么多是否正常?问题二:针对二甲基甲酰胺的测试方法,大家还有什么推荐的吗?有无标准方法?
六甲基二硅胺烷和三甲基氯硅烷作为硅甲基化试剂与醇的反应机理! 另外,六甲基二硅胺烷可以与醇反应,为什么还要加入三甲基氯硅烷?三甲基氯硅烷不是也可以与醇反应吗?请指教!







 我要推广仪器
我要推广仪器
 下载APP
下载APP