如题,现单位用到乙酸铁、乙酸铜、乙酸钴原料,但目前没有其检测方法,不知哪位大侠提供一下,谢谢了
谁有乙酸酮的分析方法啊!!~~急!!!!
有一家大型玻璃纤维拉丝和纺丝公司。拉丝车间主要污染物为给化纤丝上油的浸润剂。浸润剂的成分主成为聚酯树脂、环氧树脂、聚乙酸乙烯酯等乳液,各种表面活性剂和硅烷偶联剂等,均为乳白色水溶性化学试剂。浸润剂主要在配制反应釜中制备,配制好后输入贮罐,贮罐再输入到循环罐,循环罐再输入到每个炉台的单丝涂油器,在整个过程中都会有丙酮和乙酸的挥发。那么,我们国家有没有排放丙酮和乙酸的国家标准?排放限值(以ug/m3或mg/m3为单位)是多少?这家企业有了丙酮和乙酸的监测外,还要监测非甲烷总烃或VOCs吗?
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
职业卫生中的乙酸是用丙酮解析,但是在做标曲时丙酮和乙酸没分开,FID,WAX的柱子,请问怎样才能让它们俩分开呢
我需要测微生物发酵产生的有机酸,包括甲酸乙酸,乳酸、丙酮酸。求助大家的仪器参数建议。色谱柱的选择,已经流动相等。如果其他方法也可以测这几种物质,大家也可以给点建议。谢谢
[align=center][font='方正小标宋简体'][size=29px]巧用乙酸除铜绿[/size][/font][/align][font='仿宋_gb2312'][size=21px]我们实验室的赛默飞的[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url]购于2016年,已经使用7年,我来到实验室的时候发现上面已经长了不少绿色的物质,老师们说这个不影响实验结果,但是我想让事情是它本来该有的那个样子,所以我想试着把它清洗干净。[/size][/font][font='仿宋_gb2312'][size=21px]第一步,我分析这个绿色的物质可能是铜绿,因为我觉得这个大块金属是铜制的,查询了仪器说明书,确定是铜制的金属件后,我大胆认为是碱式碳酸铜,化学式为Cu[/size][/font][font='仿宋_gb2312'][sub][size=21px]2[/size][/sub][/font][font='仿宋_gb2312'][size=21px](OH)[/size][/font][font='仿宋_gb2312'][sub][size=21px]2[/size][/sub][/font][font='仿宋_gb2312'][size=21px]CO[/size][/font][font='仿宋_gb2312'][sub][size=21px]3[/size][/sub][/font][font='仿宋_gb2312'][size=21px]。[/size][/font][font='仿宋_gb2312'][size=21px]第二步,确定了是铜绿后,在网上查询了如何去除铜绿,有的说用乙酸和食盐;有的说用柠檬;有的说用白醋和面粉;有的说用铜清洁剂。考虑实验室已有的试剂,我打算使用纯乙酸进行清洗。首先,乙酸的酸性比碳酸强,可以与铜绿反应;其次,乙酸酸性弱,不与铜单质反应,不会腐蚀器件。[/size][/font][font='仿宋_gb2312'][size=21px]第三步,准备好乙酸、去离子水、棉签、擦镜纸。用棉签充分浸泡乙酸先简单的把有铜绿的部分全部涂抹一遍(乙酸的刺激性较强,需要戴好口罩),等一分钟,就可以开始用力擦,最不容易清洗的位置是那两道沟,用剪刀把擦镜纸剪成小块,对折两次,把回形针掰直,顶着蘸有乙酸的擦镜纸来回擦拭,铜绿完全去除后,就用去离子水润湿整张擦镜纸,反复擦拭乙酸涂抹过的区域,重复五次保证乙酸完全清除,整个过程用时90分钟左右,下面是对比图。[/size][/font][img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309010940432703_8417_5814833_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/09/202309010940433780_4938_5814833_3.jpeg[/img][font='仿宋_gb2312'][size=21px]这件事让我更加理解什么是知易行难,从我看到铜绿到开始行动去除铜绿,大约有两个月的时间,从我动手去除铜绿到清洗完毕,只用了不到两个小时的时间,知道与做到有一条巨大的鸿沟,回首工作中的点点滴滴,一道道的沟也记不清是何时走过,只是未来的路上仍不平坦,让我们身披坚强的盔甲,跨上勇气的战马,手执行动的利剑,前进四,出发![/size][/font]
高效液相测甲酸,乙酸,乳酸,丙酮酸,希望各位前辈提供一下技术参数的支持,诸如色谱柱的选择,流动相。可以用其它方法测也可以,感谢大家的建议
超高效液相色谱测甲苯,乙酸乙酯,丙酮的条件
如何将丙酮、乙酸甲酯、石油醚分开???
分离活性炭管中苯、丙酮、乙酸乙酯,FID检测器,FFAP柱(30*0.32*0.50),用二硫化碳做溶剂,请问能用用同一个条件:进样口温度150℃,检测器温度180℃,柱温40℃,以5℃/min升至80℃。把物质分离出来吗?谢谢
用乙醇一步法制乙酸乙酯的时候会产生丁酮难以分离~想问一下有什么好的方法介绍一下么?[em0808]
用气相色谱测定空气中的乙酸,用硅胶管采集后,用丙酮解吸,在做解吸效率的时候,低浓度的只有40%左右,高浓度也只有70%左右我的方法是:割开硅胶管的一端,然后用微量注射器注入用丙酮稀释过的乙酸标液,乙酸含量为50ug,100ug,400ug,然后封口,放置一夜,用2ml丙酮解吸请问各位老师我的方法对吗,还有没有更好的测解吸效率的方法呢?谢谢大家!
用气相色谱测定空气中的乙酸,用硅胶管采集后,用丙酮解吸,在做解吸效率的时候,低浓度的只有40%左右,高浓度也只有70%左右我的方法是:割开硅胶管的一端,然后用微量注射器注入用丙酮稀释过的乙酸标液,乙酸含量为50ug,100ug,400ug,然后封口,放置一夜,用2ml丙酮解吸请问各位老师我的方法对吗,还有没有更好的测解吸效率的方法呢?谢谢大家!
我一向认为乙酸乙酯卡尔费休滴定用普通试剂就可以了,有个羰基不等于醛酮.RdH的应用手册上也说直接滴定就可以了。但是梅特勒和万通的人都说要用醛酮试剂,有道理吗?
我最近在准备一个甾体原料药的申报资料,资料中要求做有关溶剂的残留的项目。在制备过程中用到了比较大量的乙酸,请问如何测定乙酸在一个甾体原料药里的溶剂残留?
我现在准备做 丙酮(分析纯试剂)、乙酸乙酯(分析纯试剂)的含量测定和水分测定,我现有的色谱柱不适用,根据国标:GB/T 686 2008 (化学试剂 丙酮):GDX-104[0.180mm—0.154mm,(80目—100目)]或选用Porapak Q[ 0.180mm—0.154mm,(80目—100目)]GB/T 12589 2007 (化学试剂 乙酸乙酯): 10%聚乙二醇己二酸酯涂于石油醚经浸泡、丙酮洗涤过的401有机担体[0.18mm—0.28mm,(60目—80目)]PS:都是填充柱大家做过同时测丙酮和水分、乙酸乙酯和水分的测定吗,什么样的柱子同时适合这两种测试呢?岔下话题:TCD检测器只能用填充柱么?
甲醛测试显色剂乙酰丙酮有颜色如何处理(因为乙酸铵变潮)?不知道大家是否有碰到过该情况,乙酸铵很容易变潮,然后配制处理的乙酰丙酮有颜色,不能使用。。。请问该如何处理?
测微生物代谢产物 甲醇、甲酸、丙酮酸、乳酸、乙酸、乙醇可以用什么检测?实验室有[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]、GC-MS。 测这些物质标曲怎么做了?浓度大概ppm级。标液溶剂一般是甲醇,那么我的甲醇又该如何检测了? 拜托各位前辈了
谁有盐酸吗啉胍和乙酸铜的检测方法
[size=3][font=SimSun]都是按国标做的,按理说不难实现,但做出来的峰惨不忍睹:GB/T 12589-2007 化学试剂 乙酸乙酯 :色谱条件与系统适用性试验 检测器:热导检测器;载气及流速:氢气,60ml/mim;柱长:2m;[color=#232323]固定相:10%聚乙二醇己二酸酯涂于经石油醚浸泡、丙酮洗涤过的401有机担体[0.18mm~0.28mm(60目~80目)],于150℃老化4h[/color][color=#333333];汽化室温度:170℃;柱温:120℃;检测器温度:150℃;各[/color]组分相对主体的相对保留值:r水,乙酸乙酯=0.18,r甲醇,乙酸乙酯=0.27,r乙醇,乙酸乙酯=0.42,r乙酸甲酯,乙酸乙酯=0.72。[color=#232323][/color]测定法 吸取本品8μl,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],记录色谱图;按峰面积归一化法计算,含乙酸乙酯([color=#333333]C[sub]4[/sub]H[sub]8[/sub]O[sub]2[/sub][/color][color=#333333])[/color][/font][/size][size=3][font=SimSun]不得少于99.5%。GB/T 686-2008 化学试剂 丙酮 :色谱条件与系统适用性试验 检测器:热导检测器;载气及流速:氢气,40ml/mim;柱长:2m;固定相:GDX-104[0.180mm~0.154mm(80目~100目)]或选用Porapak Q[0.180mm~0.154mm(80目~100目)][color=#333333];汽化室温度:170℃;柱温:130℃;检测器温度:150℃;[/color]组分相对主体的相对保留值:r水,丙酮=0.13,r甲醇,丙酮=0.30,r乙醇,丙酮=0.75。测定法 吸取本品3μl,注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],记录色谱图;按峰面积归一化法计算,含丙酮([color=#333333]C[sub]3[/sub]H[sub]6[/sub]O[/color][color=#333333])[/color]不得少于98.0%。按理说,都很好做的啊,各种温度条件都试验了,没见大的改善,不知道哪里出问题了...[font='Times New Roman'] [/font][/font][/size]
最近 在做 丙酮 石油醚60-90 乙酸乙酯 的残留检测但是由于石油醚组分复杂 在分离时 石油醚的 中的一个小峰 总是与 丙酮峰重合高手有没有好办法解决下 使用的是DB-624柱
哪位高手知道,测试有机酸如,苹果酸,乙酸,草酸,丙酮酸,甲酸等用什么柱子啊和流动相啊?
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
我要分析发酵产物中以下几种产物:丙酮、乙醇、丁醇、乙酸和丁酸。用的是“10% Carbowax-20 M, 0.10% H3PO4,support 80/100Chromosorb WAW” 玻璃填充柱和FID检测器。 因为发酵的底物中有一定浓度的糖类物质,所以采取别人的建议在衬管(liner)中填充了石英棉以阻止糖类物质进入柱子,否则糖类物质会很快的污染衬管而出现奇怪的峰。现在发现另外一个问题:那就是乙酸和丁酸总有残留,也就是说在测完一个样后,如果再测一个水样,会出现乙酸和丁酸的峰,而且峰面积比较大。不知道是乙酸和丁酸残留在柱子中,还是liner中或者其他什么地方?是否因为温度不够高?用的方法:进样口温度 225 oC;检测器温度:225 oC;柱温箱(oven)初始温度 40 oC,ramp 1: 40 oC/min for 3min, 最终温度 200 oC for 7 min; 不知有没有人有类似经历,可以给出您的建议解决这个问题?多谢!
有一个物料测试里面的4个溶剂,其中乙酸,乙酸乙酯,dmf用一种色谱条件,采用直接进样。其中丙酮采用顶空进样。是因为这两类溶剂的沸点有差异,才分别测试的吗
1、外观的测定 目测。 2、色度的测定 按GB3143规定进行测定(参见GB605-88《化学试剂色度测定通用方法》) 比色管:容量100ml。 3、乙酸含量测定 3.1 原理 以酚酞为指示剂,用氢氧化钠标准溶液中和滴定,计算时扣除甲酸含量。 3.2 试剂和溶液 氢氧化钠标准溶液:c(NaOH)=1mol/L; 酚酞指示剂:5g/L乙醇溶液,溶解0.5g酚酞于100ml的95%(V/V)乙醇中,并用4g/L氢氧化钠溶液中和至微粉红色。 3.3 仪器 锥形称量瓶(容量3ml);碱式滴定管(容量50ml)。 3.4 测定步骤 用锥形称量瓶称取约2.5g试样,准至0.0001g,将称量瓶放入已盛有50ml不含二氧化碳蒸馏水的250ml锥形瓶中,并将称量瓶盖摇开,加0.5ml酚酞指示剂,用氢氧化钠标准溶液滴定至微粉红色,保持5s不退色为终点。 3.5 计算 乙酸含量X1(%)按式(1)计算: X1=(c*V*0.06005)/m*100-1.305X2 (1) 式中 c——氢氧化钠标准溶液浓度,mol/L; V——试样消耗氢氧化钠标准溶液体积,ml; m——试样质量,g; 0.06005——与1ml氢氧化钠标准溶液[c(NaOH)=1.000mol/L]相当的以克表示的乙酸质量; X2——甲酸质量百分含量,%; 1.305——甲酸换算为乙酸的换算系数。 计算时,对标准溶液的标定与使用的温差按GB601的规定进行校正。 3.6 允许误差 两次平行测定结果差值不大于0.15%,取其算术平均值为试验结果。
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做VOC,用的WAX柱子60米长,内径0.32mm,膜厚1um,用的是标液是甲醇溶剂,热脱附进样,分流比40:1,初温35℃,8℃/min升至80℃保持2分钟,10℃/min升至115°保持2分钟,12℃/min升至160℃保持3分钟。无论条件怎么优化,都不能将乙酸乙酯,甲醇,丁酮这三个峰完全分离开,有人能提供一下优化的方法吗?[img]https://ng1.17img.cn/bbsfiles/images/2019/04/201904032334478103_9384_3416992_3.png[/img]
用茚三酮法测定氨基酸总量的时候先要配制乙酸-乙酸钠缓冲液,实验指导派介绍的是先称45.5克乙酸纳溶于100ml水中,加热溶解蒸发至60ml,冷却后加30ml乙酸,在用水定容于100ml容量瓶中.但我们在具体操作中却怎么也做不到,先是加热够冷却,但发现烧杯壁上附着厚厚一层药品,并且容量瓶里的溶液很快变成了糨糊,30ml乙酸和10ml的水根本冲不干净,误差太大.第二次干脆在还温着的时候就倒进容量瓶,但效果还是和第一次差不多.第三次是加热后趁热加乙酸进去,但还是那样.不知道别人做的时候有没有这种情况,是怎么解决的?我急啊~~~~~~~~~~~~~~~~~~```[em16] [em49]
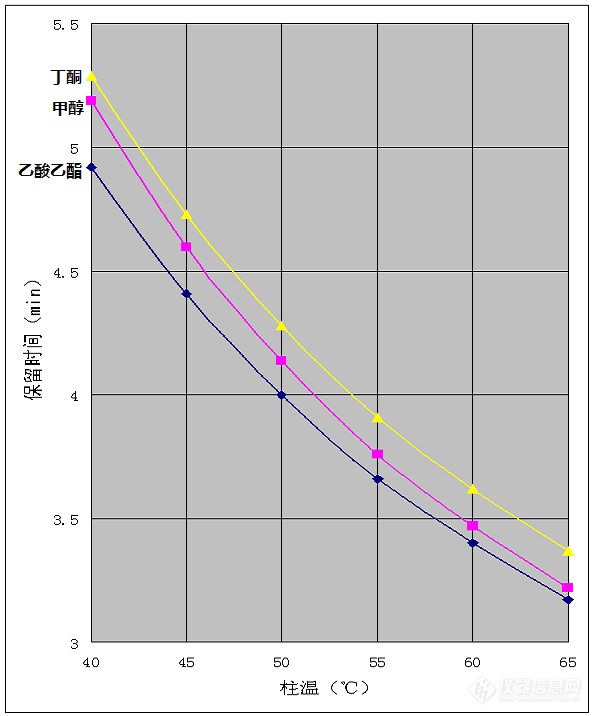
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。







 我要推广仪器
我要推广仪器
 下载APP
下载APP