甲醇氨和磷酸甲醇作为溶剂有什么作用?
请教在用石默炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]做元素Pb的时候使用了磷酸二氢氨(GR),空白进样吸光度达0.2左右,如果不用磷酸二氢氨时,吸光度为0.018左右,是不是说该优级纯磷酸二氢氨质量有问题?哪里产的GR磷酸二氢氨做LEAD合适?
求一篇聚氨酯预聚物制造中的有效阻聚剂—正磷酸的学术论文,不知道为什么公司的网关于这个论文的网站都打不开,有哪位老师有这篇论文的,麻烦发我一下,O(∩_∩)O谢谢

10,抽取5个版友);中奖名单:大川之子,纵横四海(注册ID:chuangu120)莫名其妙(注册ID:moyueqiu)吕梁山(注册ID:shih20j07)zengzhengce163(注册ID:zengzhengce163)m3071659(注册ID:m3071659)http://ng1.17img.cn/bbsfiles/images/2016/10/201610131507_613976_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/10/201610131507_613977_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================格列喹酮片方法:HPLC基质:标准溶液应用编号:101461化合物:格列喹酮固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil C18(2) 5u 150 x 4.6mm样品前处理:【有关物质】 取本品细粉适量,精密称定,加流动相溶解并稀释制成每1 ml中约含2 mg的溶液,滤过,滤液作为供试品溶液;精密量取1 ml,置100 ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。色谱条件:检测波长:UV 310 nm 流动相: 磷酸二氢铵溶液(取磷酸二氢铵1.725 g,加水300 ml使溶解,用磷酸调p H至3.5±0.05)-乙腈(3:5) 洗脱方式:等度 进样量:20 ul文章出处:P813关键字:格列喹酮片,2010版中国药典,HPLC,有关物质,钻石二代,Diamonsil C18(2)谱图:有关物质http://www.dikma.com.cn/Public/Uploads/images/geleikuitong-dz.GIFhttp://www.dikma.com.cn/Public/Uploads/images/geleikuitong-gs.GIF图例:1. 格列喹酮
GB/T 22942-2008 蜂蜜中头孢唑啉、头孢匹林、头孢氨苄、头孢洛宁、头孢喹肟残留量的测定 液相色谱-串联质谱法
尝试过用磷酸,不行~

鸡肉中喹诺酮类药物残留的检测一、方法原理 鸡肉中残留的氟喹诺酮类药物(环丙沙星、达氟沙星、恩诺沙星、沙拉沙星)用磷酸盐缓冲液提取,经HLB固相萃取柱净化,流动相洗脱后。用液相色谱-荧光检测器测定,外标法定量。二、仪器设备 安捷伦1100高效液相色谱仪(配荧光检测器) 涡旋振荡器 冷冻离心机 固相萃取装置三、实验部分 ★试剂的配制 1.磷酸盐缓冲液:取磷酸二氢钾6.8g,加水溶解并稀释500ml,用5.0mcl/ml氢氧化钠溶液调节(PH=7.0) 2.磷酸/三乙胺溶液(0.05mcl/l)取85﹪磷酸3.4 ml,用水稀释至1000ml,用三乙胺调节PH=2.4. 3.标准工作液的配制 3.1 分别称取盐酸环丙沙星、甲磺酸达氟沙星、恩诺沙星、盐酸沙拉沙星对照品各约25mg,精密称定,分别于25ml棕色容量瓶中,用0.03mcl/l氢氧化钠溶液稀释成浓 度 ,分别为1mg/ml的储备液。(达氟沙星储备液浓度配制成0.2mg/ml) 3.2混合标准中间液 分别量取1mg/ml(0.2mg/ml)的储备液各0.1ml于50ml容量瓶中,用流动相稀释成浓度为2ug/ml(0.4ug/ml)的混合标准中间液。★方法步骤 1.质量控制样品的制备 1.1空白样品 取空白试样,与待测样品同步处理。 1.2添加样品 取空白样品,添加2ug/ml(0.4ug/ml)的混合标准中间液0.1ml,制的浓度为100ug/kg的阳性添加试料,与待测样品同步处理。 2.提取http://ng1.17img.cn/bbsfiles/images/2015/11/201511271155_575220_2315779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015112711410717_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015112711395737_01_2315779_3.png 2.1取匀浆的试料2±0.02g,置50ml离心管中,加磷酸盐缓冲液10.0ml,涡旋混合,中速震荡5min,14000r/min离心10min,倾取上清液。(磷酸盐缓冲液:取磷酸二氢钾6.8g, 加水溶解并稀释500ml,用5.0mcl/ml氢氧化钠溶液调节PH=7.0) 2.2用磷酸盐缓冲液10.0ml重复提取一次。合并两次上清液,备用。 3.净化 3.1 固相萃取柱依次用甲醇2ml,磷酸盐缓冲液2ml预洗。 3.2 取上述备用液体5.0ml过柱。 3.3 用水2ml洗柱,挤干。 3.4用流动相2.0ml洗脱,收集,挤干,涡旋混合。过0.22um微孔滤膜,供上机测定。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271156_575226_2315779_3.png 4.系列标准工作液的制备 精密量取混合标准中间工作液,用流动相稀释成浓度为5、10、20、50、100、200、500ng/ml的系列标准工作液。★仪器色谱条件 色谱柱:C18(4.6×150mm 5um); 柱温:30℃; 流动相:0.05mcl/l磷酸溶液/三乙胺-乙睛(87+13); 流速:1.0ml/min; 进样量:20ul; 检测波长:激发波长280nm;发射波长450nm.★结果分析 1、线性范围 取系列标准工作液进样,以峰面积为纵坐标,以药物浓度为横坐标绘制标准工作曲线。结果表明(见表1),在系列标准工作液浓度范围内,喹诺酮类药物峰面积与浓 度呈良好的线性关系。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271156_575224_2315779_3.png 2.准确度的测定 称取同一来源的空白鸡肉样品3份,每份2.0g,分别添加2ug/ml(0.4ug/ml)的喹诺酮类药物溶液0.1ml的阳性样品,按本方法测定,计算回收率,见表2http://ng1.17img.cn/bbsfiles/images/2015/11/201511271200_575228_2315779_3.pnghttp://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif四、结论 本文建立鸡肉中喹诺酮类药物的液相色谱-荧光测定的方法。鸡肉样品经磷酸盐缓冲液提取后,经固相萃取柱净化后,采用液相-荧光检测器法测定,外标法定量,经检测被测样品没有检测出含喹诺酮类药物残留成分,阳性添加样品的回收率为73.1%-84.5%,相对标准偏差为1.8%-5.4%(n=3).本方法系列标准工作液浓度范围内,喹诺酮类药物峰面积与浓度呈良好的线性关系,准确度好、适用于鸡肉中喹诺酮类药物残留的检测。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271156_575222_2315779_3.png 空白鸡肉样品的色谱图http://ng1.17img.cn/bbsfiles/images/2015/11/201511271156_575223_2315779_3.png http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif100ug/kg空白鸡肉添加试样中氟喹诺酮类药物色谱图http://ng1.17img.cn/bbsfiles/images/2015/11/201511271156_575225_2315779_3.bmp 20ng/ml 标准溶液中氟喹诺酮类药物色谱图
同志们,BOD5化验中的磷酸盐需要一个药品叫磷酸氢二钾,潮解了怎么办
我现在检测牛肉中氟喹诺酮类物质的含量,对照浓度为0.05μg/ml,标液为4μg/ml,取2g牛肉加入100μl的标液再加入20ml磷酸盐缓冲溶剂,振荡离心,取5ml上清液过HLB的固相萃取柱,最后用2ml流动相洗脱上机检测,回收率特别低,怎么才可以提高回收率呢?
大伙能给我解答一下,正磷酸盐,总无机磷酸盐和总磷的区别吗?还有氨氮和总氮
如题,有一肥料磷酸二氢钾样品,我按HG2321-92的标准来进行磷酸二氢钾含量的检测(第一法喹钼柠酮重量法),测得其含量为98.5%,而氧化钾的含量按HG2321-92的标准(四苯硼酸钠重量法)来检测只有31。4%,可是如何按磷酸二氢钾的含量来换算氧化钾的质量分数的话:98.5*94/136.1/2=34.0啊,34.0与31.4的含量相差已经超出了实验室的允许误差啊,而且该样品磷酸二氢钾含量合格,而氧化钾的含量却不合格,也说不过去啊,怀疑是氧化钾含量被分析低了,或者是磷酸二氢钾分析出错了,同时我有将分析纯的化学试剂磷酸二氢钾同时带入试验,而分析的结果是磷酸二氢钾的含量与氧化钾的含量符合二者之间的换算关系啊,为什么一到肥料磷酸二氢钾时,这种换算关系就不存在了??
最近一个月,使用同一批次配置好的喹钼柠酮分析磷铵产品中五氧化二磷含量,重量法结果比容量法结果高出近一个品位,使用PT级磷酸二氢钾试验后发现,容量法结果准确。按理说重量法是国标,不应不准的啊,换用其他实验室的喹钼柠酮沉淀剂,结果与容量法吻合,不知道我们自己配制的沉淀剂出现了什么问题???
磷酸在湿法消解中常用,但是在微波消解中不常用,有一定风险。那是否可以在微波消解中使用呢?使用量该如何控制?
买到的磷酸都是用塑料瓶装的。热的磷酸似乎有碱性?配制的磷酸溶液(比如1:1的磷酸溶液)可以用玻璃试剂瓶存放么?
有没有哪位大侠做过氟喹诺酮类药物的残留检测啊,我是使用WATERS的HLB小柱,甲醇-水活化,50mM乙酸氨溶解样品,上样,用2%甲酸水,甲醇淋洗,然后5%甲醇氨洗脱,加了内标校正,回收率高达130-190%,很吓人请大侠们给俺点建议,咋办呢? [em61] [em61]
磷酸有较低的蒸气压,在0.8MPa时温度可达240 ℃。热HP04 适用于消解那些用HCl消解时会使某些特定痕量组分挥发损失的铁基合金,磷酸还可溶解铬矿、氧化铁矿、铝炉渣等。你是否在微波消解中使用过磷酸?当你在使用磷酸消解样品的时候一般是与其它酸组合使用的吧?具体用于哪类样品的消解?
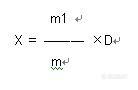
标准简介:GB/T 8381.10-2005 饲料中磺胺喹恶啉的测定高效液相色谱法 本标准规定了以高效液相色谱(HPLC)仪测定饲料中磺胺喹恶啉的方法。本标准适用于配合饲料、浓缩饲料和添加剂预混合饲料中磺胺喹恶啉的测定,最小检测浓度为5.0 mg/kg。磺胺喹恶啉属磺胺类抗生素兼有抗球虫作用,广泛用于养禽业,其口服后吸收迅速,但排泄缓慢,残留在组织器官及鸡蛋中时间长,人食用后,对人体产生耐药性等副作用。世界卫生组织(WHO)规定动物组织、奶的最高残留限量(MRL)值为100μg/kg。日本等国规定肉鸡中不得检出。因此,该药的使用应严格控制用药期及用药量,以保证产品的安全及对外贸易畅通。英国兽药典记载,磺胺喹恶啉纯品为黄色粉末,几乎无味,不溶于水,溶于甲醇、乙醇,易溶于碱性溶液。目前,饲料中磺胺喹恶啉的检测方法美国AOAC标准是采用分光光度法,该法要求含阿散酸及不含阿散酸的饲料采用两种不同的测定方法。而国内饲料目前添加组分较复杂,检测者对饲料情况是未知的,难以确定用哪一个方法;且该方法中需要重氮化试剂做偶合反应,其毒性大,对操作者及环境 都会造成危害。国标GB/T 8381.10中,规定了采用高效液相色谱法测定饲料中磺胺喹恶啉。方法简介如下:1.测定方法1.1原理 用甲醇水溶液提取饲料中的磺胺喹恶啉,离心,过滤,在HPLC仪上分离,紫外检测器240nm处测定。1.2 试剂和溶液以下所用的试剂,除特别注明外均为分析纯试剂;水为蒸馏水,色谱用水符合GB/T6682规定的一级水。1.2.1 磺胺喹恶啉标准品:含磺胺喹恶啉(C14H12N4O2S) 95.0%。1.2.2 甲醇:色谱纯。1.2.3 磷酸盐溶液:取磷酸二氢钾3.40g和磷酸氢二钾5.71g,加水溶解并稀释至1000mL。1.2.4 磺胺喹恶啉标准溶液:准确称取磺胺喹恶啉(4.1)50mg,溶于甲醇并稀释成0.1mg/mL的储备液,置4℃冰箱中避光保存,有效期1个月。临用前,取此储备液用水稀释成适当浓度的标准工作液。1.2.5 提取液:甲醇100mL+水50mL1.3 仪器1.3.1 实验室常用仪器设备1.3.2高效液相色谱仪(配紫外检测器)1.3.3 分析天平:感量为0.0001g和0.001g1.3.4 旋涡振荡器1.3.5 离心机:4000r/min1.3.6针头过滤器:备孔径为0.45μm 微孔滤膜1.4 分析步骤1.4.1 提取 称取5g试样,精确至0.001g,加入提取液(4.5)50mL,旋涡振荡器混匀,超声水浴中提取15min,中间取出摇动1次,然后4000r/min离心5min,静置,取上清液过0.45μm滤膜,供液相色谱测定。1.4.2 标准曲线的制备 准确吸取储备液适量,用水或流动相稀释成浓度分别为0.10、0.50、1.00、2.00、10.0μg/mL的磺胺喹恶啉标准溶液,做出标准曲线。 1.4.3 测定1.4.3.1 色谱条件 色谱柱:C18柱 柱长150mm,柱内径4.6mm,粒度5μm或性能相当者。 流动相:磷酸盐溶液75mL+甲醇25mL,用前过0.45μm滤膜,并超声脱气。 流速:1mL/min。 检测波长:240nm。 进样量:10~20μL。1.4.3.2 定性与定量 根据标准品的保留时间定性,定量由标准曲线或单点校准。1.4.4 结果的计算 每千克试样中含磺胺喹恶啉的质量按下式公式计算: http://ng1.17img.cn/bbsfiles/images/2010/11/201011131559_259214_1620630_3.jpg x——每千克试样中磺胺喹恶啉的质量(mg); m1——色谱峰面积对应的磺胺喹恶啉的质量(μg); D——稀释倍数; m——所称样品的质量(g)。 平行测定结果用算术平均值表示,保留至小数点后1位。
玻璃中的氟含量较高,氟元素含量在20-50wt%之间,氧在1-20wt%之间。样品所含的主要元素有F O P Al Li Na K Mg Ca Sr Ba Y,不同样品所含元素稍有差异。目前试了一下,元素分析仪由于样品中含磷酸盐和氟所以测不了。氢氧氮分析仪由于氟浓度太高了,高温下跟燃烧管反应,所以也测不了。请问氟磷酸盐玻璃中的氧元素含量检测还有哪些可用的方法?
样品需要稀释倍数做BOD5在稀释水里加磷酸盐和氯化铁等溶液,用稀释水做空白需要加磷酸盐和氯化铁等溶液吗?我今天准备做葡萄糖和谷氨酸标准样品,稀释水需要加那几种溶液吗?
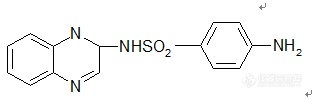
【中文名称】磺胺喹喔啉;2-(对氨基苯磺酰胺基)喹喔啉【英文名称】sulfaquinoxaline【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203021946_352107_1855403_3.jpg 【熔点(℃)】248~255【性状】 淡黄色结晶粉末。【溶解情况】 几乎不溶于水、乙醇、丙酮,溶于碳酸钠和氢氧化钠水溶液。【用途】 广泛用于抗球虫病,对鸡巨型艾美耳球虫、布氏艾美耳球虫和堆形艾美耳球虫作用最强,与氨丙啉合用抗球虫效果更佳。禁用于产蛋鸡。【制备或来源】 由邻苯二胺鱼氰化钠、甲醛在酸性介质中反应制得N-氰甲基啉本二胺,然后与氢氧化钾环合成2-氨基-3,4--二氢喹喔啉,最后,与乙酰胺基苯磺酰氯缩合、水解而得。【其他】 其钠盐为无定型粉末,易溶于水。【生产单位】略
在做氨氮和总氮的试验中,出现了氨氮比总氮高的现象,请问,这是什么原因?同时,在做磷酸盐和总磷的试验中,也遇到这样的问题,磷酸盐比总磷高,真是糊涂了。难倒是光度计出现了问题吗???
2005《中国药典》头孢氨苄含量测定项下: 色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;以水-甲醇-3.86%醋酸钠溶液-4%醋酸溶液(742:240:15:3)为流动相;检测波长为254nm。头孢氨苄干混悬剂、头孢氨苄片、头孢氨苄胶囊和头孢氨苄颗粒含量测定方法同头孢氨苄。 文献报道的方法(HPLC法),试验条件除药典的水-甲醇-3.86%醋酸钠溶液-4%醋酸溶液系统外,其它的如: 周嘉等用HPLC法测定头孢氨苄胶囊的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱为Spherisorb C18柱(5μm,4.6mm×250mm);流动相为0.025mol/L磷酸溶液(用三乙胺调pH3.0±0.1)-乙腈(88:12);流速为0.9ml/min;检测波长为240nm;柱温为室温。 王建宁等用HPLC法测定头孢氨苄片的含量。仪器:日立L-6200A高效液相色谱仪。色谱柱为C18(250mm×4.6mm,5μm);流动相:0.04mol/L磷酸溶液(用三乙胺调pH3.0)-乙腈(75:25);流速:1mL/min;检测波长:240nm;柱温:室温。 程成等用HPLC法测定头孢氨苄胶囊的含量。Waters色谱系统,色谱柱采用Nova Pak ODs柱(4μm,4.6mm×250mm);流动相为0.01mol/L醋酸铵溶液(冰醋酸调pH4.0)-甲醇(70:30);流速:0.8ml/min;检测波长262nm;柱温25℃。 周静安等用HPLC法测定头孢氨苄胶囊的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱为Shim-pack CLC-ODS柱(150mm×6mm);流动相为甲醇-水(70:30);流速为1.0ml/min;检测波长为262nm。 邓永辉等HPLC法测定头孢氨苄的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱:250mm×4.6mm 不锈钢柱,填料为Hypersil ODS;磷酸二氰钾溶液(0.01mol/L)-乙氰-甲醇(90:8:2),并用磷酸溶液调节PH 至4.5±0.1;检测波长:254mm。 崔慈等用HPLC法测定头孢氨苄的含量。仪器:上海伍丰LC-100液相色谱仪。色谱柱:150mm的C18柱子,以0.025mol/L磷酸溶液(用20%NaOH调节PH至3左右)-乙腈88:12为流动相:检测波长为235nm。如果各位还有不同方法,欢迎发布交流,谢谢!
我使用石墨炉氘灯扣背景做食品样,使用磷酸氢氨做基体改进剂,可是背景扣得不理想,是否是加热程序的问题?如果是,如何设置妥当?
进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。

10,抽取5个版友);中奖名单:zengzhengce163(注册ID:zengzhengce163)梧桐(注册ID:mengzhou)m3071659(注册ID:m3071659)999youran(注册ID:999youran)sixingxing(注册ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/05/201605121503_593117_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/05/201605121503_593118_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================动物组织中喹诺酮类药物( 沙星类药物) 的测定方法:SPE基质:动物组织应用编号:101078化合物:马波沙星; 氧氟沙星; 诺氟沙星; 恩诺沙星; 环丙沙星; 帕珠沙星; 双氟沙星; 沙拉沙星; 加替沙星;司帕沙星;恶喹酸;萘啶酸;氟甲喹固定相:ProElut PXA色谱柱/前处理小柱:ProElut PXA 150mg / 6ml 30/pkg样品前处理:1、提取 将2 g 试样置于50 mL 塑料离心管中,加入2 g 无水硫酸钠和20 mL 乙腈,10000 rpm 下均质1min,4000 rpm 下离心5 min,收集上清液于50 mL 比色管,20 mL 乙腈重复提取一次,合并上清液。用10 mL*2 正己烷对提取液进行脱脂,正己烷弃去。将乙腈层减压蒸馏至近干,6 mL 5% 氨水溶液分三次溶解,待净化。 2、净化 ProElut PXA 150 mg/6 mL (Cat.#68304) a 活化: 6 mL 甲醇活化、6 mL 水平衡; b 上样: 将待净化液加入小柱,流出液弃去; c 淋洗: 依次用6 mL 水、6 mL 甲醇淋洗小柱,流出液弃去; d 洗脱: 6 mL 2% 甲酸甲醇溶液洗脱,收集洗脱液; e 重新溶解:30 ℃ 下将洗脱液减压蒸馏至近干,1 mL 流动相溶解,微孔滤膜过滤后供HPLC 分析 注:e 步骤是供参考的,使用者可根据自己使用的分析仪器进行调整。色谱条件:色谱柱:Diamonsil C18(2) 250 x 4.6 mm ID, 5 μm (Cat. #99603) 流速:1 mL/min 进样量:20 μL 检测器:UV 280 nm 柱温:35 ℃ 流动相:A :甲醇,B :0.2% 磷酸水溶液,梯度 梯度设置 T 0 25 40 42 50 A 22 33 65 22 22 B 78 67 35 78 78 注:对于氟喹诺酮类药物分析,紫外检测器并非最适宜的检测器,如果条件具备,分析工作者最好使用荧光检测器或质谱检测器进行检测。其中T为时间(min)文章出处:P113关键字:动物组织,沙星类药物,SPE,ProElut PXA,马波沙星; 氧氟沙星; 诺氟沙星; 恩诺沙星; 环丙沙星; 帕珠沙星; 双氟沙星; 沙拉沙星; 加替沙星;司帕沙星;恶喹酸;萘啶酸;氟甲喹摘要:适用于禽、畜、水产品组织中喹诺酮类药物的检测。谱图:http://ng1.17img.cn/bbsfiles/images/2016/05/201605121000_593072_1610895_3.jpg图例:1. 马波沙星;2. 氧氟沙星;3. 诺氟沙星;4. 恩诺沙星;5. 环丙沙星;6. 帕珠沙星;7. 双氟沙星;8. 沙拉沙星;9. 加替沙星;10. 司帕沙星;11. 恶喹酸;12. 萘啶酸;13. 氟甲喹
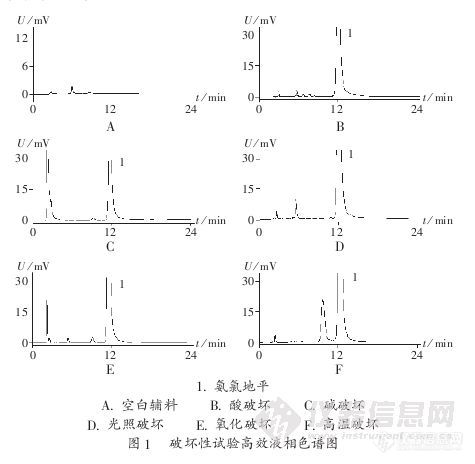
作者:秦书德 马晓伟 陈静 郭兴杰作者单位沈阳药科大学,摘 要:目的建立检查苯磺酸氨氯地平有关物质的反相高效液相色谱法。方法采用Diamonsil C18色谱柱(200 mm×4.6 mm,5μm)进行分离,流动相为甲醇-30 mmol/L磷酸二氢钾溶液(60∶40),流速1.0 mL/min,检测波长238 nm。结果所用色谱条件能很好地分离氨氯地平及其降解产物,氨氯地平质量浓度在0.71~3.57μg/mL范围内与峰面积线性关系良好。结论反相高效液相色谱法专属性强、准确、灵敏,可用于苯磺酸氨氯地平片中有关物质的检查:http://ng1.17img.cn/bbsfiles/images/2012/08/201208201631_384755_2379123_3.jpg
微波辅助萃取-GC-MS法测定玩具中磷酸三(2-氯乙基)酯 作者:蒋小良http://ng1.17img.cn/bbsfiles/images/2015/11/201511271307_575230_2904170_3.jpg 摘要:建立了微波辅助萃取-气相色谱-质谱法(GC-MS)测定玩具中磷酸三(2-氯乙基)酯含量的分析方法。考察了6种不同萃取溶剂对磷酸三(2-氯乙基)酯的萃取效率,选择了以乙酸乙酯为萃取溶剂,考察了微波辅助萃取温度与时间对萃取效果的影响,选择了最佳的微波辅助萃取条件。萃取液经浓缩后,用气相色谱-质谱仪进行选择离子监测模式下的定性及定量分析。结果表明,该方法的检出限为0.02 mg/kg,样品的加标回收率为93.6%~98.2%,相对标准偏差小于4.8%。该方法具有快速、方便、灵敏度高、定性准确等优点,适用于玩具中禁用有机磷阻燃剂磷酸三(2-氯乙基)酯含量的快速测定。 关键词:微波辅助萃取;磷酸三(2-氯乙基)酯;玩具;气相色谱-质谱法 近年来,由于溴代阻燃剂在全世界范围内逐步禁止使用,有机磷酸酯类阻燃剂作为其主要替代产品,其生产与使用得到了大幅度增长。所以,有机磷酸酯类物质也被广泛用于儿童产品(包括玩具)的阻燃整理工艺中,由此所引起的环境问题逐步引起了环境科学工作者的关注。有机磷酸酯类物质成为新型有机污染物研究又一个热点。有机磷阻燃剂化学性质很稳定,难以降解,具有生物累积性,能够侵害大脑组织,从而永久性地损伤人记忆和学习能力,并且具有致癌性,欧洲化学物质信息系统里明确将磷酸三(2-氯乙基)酯(tris(2-chloroethylphosphate), 简称TCEP)定义为致癌、生殖毒性和生态污染物。2010年1月,欧洲化学品管理局将三-( 2-氯乙基) 磷酸酯列入第二批授权物质清单;2012年,加拿大政府发布拟议法规修正案,将禁止在三岁以下儿童使用的产品(整体或部件)中使用含有磷酸三(2-氯乙基)酯的聚氨酯泡沫(PUF);欧洲报检和环境科学委员会(SCHER)就玩具中的TCEP表述科学观点,规定其限值为5 mg/kg,SCHER表示应该在玩具中禁用TECP及其同系物,此观点获得欧洲标准化消费者之声(Anec)和欧洲消费者联盟 (Beuc)的支持。为了更好的保护儿童健康和安全,欧委会接受相关组织的建议,发布2009/48/EC修订法案COM/2012/003,规定禁止在玩具中禁用TECP及其同系物;2011年7月,华盛顿生态部批准通过了《儿童产品安全法申报规则》第173-334-WAC章,该条例旨在收集相关信息,帮助政府和公众更好地了解儿童产品中潜在危险的化学品。该规则要求儿童产品生产商(包括品牌所有者和进口商)向生态部申报产品中对儿童具有高风险的化学品(CHCCs)的存在情况,其中TCEP已收录在需申报的化学品清单里。目前检测分析三-( 2-氯乙基) 磷酸酯主要方法有气相色谱法和气相色谱-质谱联用法。本文采用微波辅助乙酸乙酯萃取,气相色谱-质谱法(GC-MS)测定玩具中的三-( 2-氯乙基) 磷酸酯,对微波萃取条件进行了实验,选择了最近的萃取条件,并对该方法的精密度和加标回收率进行了系统研究,建立了玩具中三-( 2-氯乙基) 磷酸酯的快速测定方法。1、实验部分1.1 仪器和试剂 QP2010 Plus型 气相色谱-质谱仪 (日本SHIMDAZU公司);TB215D型 电子天平(美国丹佛公司);Ethos ONE型 微波消解/萃取仪(意大利Milestone 公司);EV321型 旋转蒸发仪(北京莱伯泰科仪器有限公司);ZM 200超离心粉碎仪(德国RETSCH公司);超声波发生器(上海之信仪器有限公司);21011V001R200型 氮吹仪(瑞士BUCHI公司)。磷酸三(2-氯乙基)酯(纯度≥98.5 %,DrEhrenstorfer公司),丙酮、乙酸乙酯、甲醇、乙腈、乙醇和正己烷均为色谱纯,均购自Sigma-Aldrich公司。磷酸三(2-氯乙基)酯标准储备溶液1000mg/L:精确称取磷酸三(2-氯乙基)酯标准品0. 10g,用丙酮溶解并定容至100 mL。临用前用乙酸乙酯将磷酸三(2-氯乙基)酯标准储备液稀释成50 mg/L的标准工作溶液。1.2 气相色谱-质谱条件 ( 1 )色谱条件:Rtx-5MS色谱柱( 30 m ×0. 25 mm×0. 25μm) ;升温程序: 初始温度70 ℃(保持2min),以20℃/min升至280 ℃,再以25 ℃/min升至290 ℃保持1 min; 载气: 高纯氦气; 流速: 1. 0 mL /min; 进样口温度280 ℃; 色谱-质谱接口温度:280 ℃;进样量: 1. 0μL;进样方式:不分流进样;溶剂延迟:5min。 (2)质谱条件:离子源温度230 ℃;四极杆温度:150℃;EI源:电子能量70 eV;扫描方式:全扫描;扫描范围:50~500amu。1.3 样品处理 取具有代表性的玩具样品10 g,剪碎至约为5 mm×5 mm,经超离心粉碎仪粉碎后(过1mm筛)。准确称取1 g(精确至0.001 g)样品于微波萃取罐中,加入15mL乙酸乙酯,按照选定的微波萃取条件进行萃取,萃取完成后,待萃取液冷却至室温,将萃取液转移至鸡心瓶中,并再用15mL乙酸乙酯分三次洗涤萃取残渣,合并萃取液及洗涤液,旋转蒸发(控制温度低于45℃)至近干,再用氮气吹至近干,用2mL乙酸乙酯溶解残渣并定容,再用0.2μm 滤膜过滤,滤液上气相色谱-质谱仪进行分析。2、结果与讨论2. 1 微波萃取条件的选择 影响微波辅助萃取效率的主要因素包括萃取溶剂种类和萃取条件,萃取条件主要有:萃取温度、萃取时间及萃取压力等。因此选择合适的萃取剂、萃取剂用量、萃取温度和萃取时间等,将直接影响样品中待测物萃取的效果及后续测试的结果准确度。2.1.1 萃取剂的选择 在微波辅助萃取中,选择萃取溶剂时,不仅要考虑样品中目标分析物在所选择溶剂中的溶解度,而且要考虑所选溶剂与样品基质的相互作用,以及所选溶剂对微波的吸收情况,溶剂的极性越大,对微波的吸收越强。按照样品处理程序1.3试验了二氯甲烷、乙酸乙酯、乙醇、甲醇、乙腈和正己烷6种萃取溶剂,对4种不同玩具产品进行萃取试验,实验结果见表1,由表1可见,在相同的萃取条件下,乙酸乙酯的萃取效率最高,所以实验选择乙酸乙酯作为萃取溶剂。http://ng1.17img.cn/bbsfiles/images/2015/11/201511271317_575231_2904170_3.jpg2.1.2 微波萃取温度和时间的选择 微波辅助萃取温度是萃取过程中的重要参数之一,微波温度的高低将直接影响萃取速率和效率。试验研究表明,在进行
老师们好:现在我们实验室要测试污水的:COD、 BOD5、 氨氮 、悬浮物 、PH、色度、这几个项目1、PH、色度这两个项目没有什么问题,PH就是PH计,色度就用原始的方法,试管比色法测,肯定会有些误差2、悬浮物还没有测过,但是查到国标了,好早以前的1989年的,http://simg.instrument.com.cn/bbs/images/brow/em53.gif不知道有没有更新,有更新的话老师们告诉一下3、COD BOD5 氨氮这三个项目想买仪器测,人工测太麻烦了,大概查了查,说连华的不错,大家用过吗?用过的讲解一下吧,氨氮这个项目有国标吗?有的话也麻烦老师推 荐下
这个问题困扰我好几天了,在此之前没有遇到过类似问题。初步试验下来,磷酸缓冲液中所使用的磷酸氢二钾,磷酸氢二钠都会与氯化钙反应产生沉淀,估计是磷酸氢钙或者磷酸钙。磷酸二氢钾没有发现与氯化钙反应。已经申请购买新的磷酸氢二钾和磷酸氢二钠;氯化钙是新购入的,批次很新,应该没问题。我请问一下,除了试剂本身变质,还有什么原因可能会导致像现在这样的情况?各位在做BOD培养时,有没有遇到过类似情形?如果告知,不胜感激!
赛场岁月2——畜产品氟喹诺酮残留检测比武小记 虽然是检测比武的小记,但内容绝大多数是前处理的,所以就发在这个版块,特此说明。1前言2013年9月25~27日,浙江省第四届农产品质量安全检测技能竞赛暨第二届全国检测技能竞赛浙江省预赛在杭州举行,本人有幸参加了畜产品组比武。2比赛项目本次比武的项目是鸡肉中氟喹诺酮残留检测,依据的标准是《动物性食品中氟喹诺酮类药物残留检测高效液相色谱法》(农业部1025号公告-14-2008)。比武的主要内容是取已匀浆的鸡肉样品,加标后进行前处理及净化,然后将提取液交赛事工作人员统一上机处理,拿到数据后填写原始记录上交。3赛前戏言比武前赛事组织单位提供了几个复习项目(如猪肝中β-激动剂[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测、猪肉中磺胺总量液相检测、鸡肉中氟喹诺酮类液相检测等),得知比赛项目后确实很惊讶,因为这个项目只需称取样品、匀浆、离心、上清液过柱净化即可,以至于比赛前我忍不住嘀咕了句:“考这么简单的项目?”当时一位裁判老师听闻后对我说:“小心阴沟里翻船!”事实证明裁判老师的预言不无道理!4标准相关内容《动物性食品中氟喹诺酮类药物残留检测高效液相色谱法》(农业部1025号公告-14-2008)有关提取和净化的内容如下:提取:取(2±0.05) g试料,置30 mL匀浆杯中,加磷酸盐缓冲溶液10.0 mL,10 000 r/min匀浆1 min。匀浆液装入离心管中,中速振荡5 min,离心(肌肉、脂肪10 000 r/min 5min;肝、肾15 000 r/min 10min),取上清液,待用。用磷酸盐缓冲溶液10.0 mL洗刀头及匀浆杯,转入离心管,洗残渣,混匀,中速振荡5 min,离心(肌肉、脂肪10 000 r/min 5min;肝、肾15 000 r/min 10min)。合并两次上清液,混匀,备用。净化:固相萃取柱先依次用甲醇、磷酸盐缓冲溶液各2 mL预洗。取上清液5.0 mL过柱,用水1 mL淋洗,挤干。用流动相1.0 mL洗脱,挤干,收集洗脱液。经滤膜过滤后作为试样溶液,供高效液相色谱法测定。5比赛规定处理程序考虑到比赛进度,以及仪器设备交叉使用等问题,比赛时的处理程序为:提取:取(2±0.05) g试料,置于50mL塑料离心管中,加磷酸盐缓冲溶液10.0 mL,10 000 r/min匀浆1 min。取一小烧杯用磷酸盐缓冲溶液10.0 mL洗刀头,备用。离心管中速振荡5 min,10 000 r/min 离心5 min,取上清液。用小烧杯中溶液洗离心管残渣,混匀,中速振荡5 min,10 000 r/min 离心5 min,取上清液。合并两次上清液,混匀,备用。净化:固相萃取柱先依次用甲醇、磷酸盐缓冲溶液各2 mL预洗。取上清液5.0 mL过柱,用水1 mL淋洗,挤干。用流动相1.0 mL洗脱,挤干,收集洗脱液。经滤膜过滤后作为试样溶液,供高效液相色谱法测定。6操作过程需要说明的是,比赛所需的样品、试剂均由赛事组织单位提前完成制备,设备均已处于开机状态,选手可以直接使用。6.1称取样品要求:称取(2±0.05)g样品,置于50mL塑料离心管中。操作:(1)检查天平是否处于水平位置,若否应调整至水平位置,并向裁判示意;(2)取一玻璃烧杯作盛放离心管容器,置于天平;(3)取50mL塑料离心管,做好标记(1A、2A);(4)取1A号离心管置于烧杯中,天平去皮;(5)用药匙先将鸡肉样品充分混匀,刮取适量置于离心管底部;(6)置于天平称量,并检查是否处于要求范围内,若否进行适当调整;(7)依样进行第2份样品操作。要点:(1)称量前一定要检查天平是否处于水平状态;(2)塑料离心管共取4支,2支用于盛放样品(编号为1A、2A),2支用于离心时平衡(编号为1B、2B);(3)鸡肉样品从冷冻状态取出后回温,有固液分离现象,取样前需先用药匙将样品搅拌均匀,否则实验结果平行性可能不佳;(4)取样时务必将样品置于离心管底部,不能粘在离心管壁上部。6.2加标要求:加入氟喹诺酮类混合标样0.5mL。操作:(1)取一玻璃烧杯,做好标记;(2)于烧杯内倒入适量混合标样;(3)取一吸管,做好标记;(4)吸取混合标样,对吸管进行润洗操作数次,液体放入废液缸;(5)吸取混合标样,小心放液至刻度0处,向离心管放液0.5mL;(6)依样进行第2份样品操作。要点:(1)烧杯、吸管宜做好标记,以免忙中出错;(2)吸管一定要进行润洗;(3)观察容量时刻度线要与眼睛齐平;(4)废液一定要放入指定的缸内。6.3加提取液要求:加入磷酸盐缓冲液10.0mL。操作及要点:与6.2相似。6.4匀浆要求:10000 r/min匀浆1min。操作:(1)将匀浆机刀头浸入1A号离心管液面以下,开启匀浆机进行匀浆;(2)匀浆时间控制在1min左右,时间过长对匀浆效果并无明显改善,且产生的高温反而可能导致待检成分分解;(3)匀浆完成后取走离心管,用预先准备的装有磷酸盐缓冲液10.0mL的小烧杯对刀头进行清洗,并用干净的滤纸吸干刀头上的水分。要点:(1)清洗刀头的烧杯和磷酸盐缓冲液要预先准备好;(2)匀浆速度调整至何位置要预先熟悉;(3)匀浆时要注意随时移动离心管,使匀浆充分均匀;(4)匀浆完成后应及时对刀头进行清洗,以免刀头未洗造成待检成分损失;(5)如刀头内有纤维缠绕应及时取出,并擦干水分,以免影响其他选手操作。6.5振荡要求:中速振荡(约160 r/min) 5 min。操作:(1)将完成匀浆的离心管放入振荡器;(2)开启振荡器进行振荡提取;(3)结束后取出离心管;要点:(1)离心管一定要旋紧盖子,避免提取液漏出;(2)某些振荡器利用弹簧对离心管进行固定,振荡过程中应不时观察离心管是否脱离夹具。6.6离心要求:离心(10 000 r/min 5min),取上清液。操作:(1)玻璃烧杯置于天平并去皮置零;(2)分别对离心管(编号为1A、2A)进行称重并记录;(3)分别将离心管(编号为1B、2B)放入烧杯,加水至与离心管(编号为1A、2A)相同质量;(4)将4支离心管按两两对应放入离心机中进行离心;(5)离心结束后小心取出离心管;(6)取一新的50mL离心管,小心将上清转入。要点:(1)离心前一定要选进行平衡;(2)离心结束后取离心管和转移上清时,一定要轻拿轻放;(3)盛放上清液的离心管编号,应与原离心管编号相同,提前做好标记。6.7洗残渣、振荡、离心操作:(1)将小烧杯中的磷酸盐缓冲液10.0mL倒入离心管中,并盖紧离心管盖;(2)离心管置于旋涡振荡器中涡旋1min,使残渣充分分散;(3)振荡及离心参照6.5及6.6;(4)合并两次上清液。要点:(1)小烧杯中的磷酸盐缓冲液与离心管要一一对应;(2)离心管盖一定要旋紧,否则涡旋时易漏液;(3)涡旋时需要掌握一定的角度与力度,否则效果不佳。6.8固相柱活化操作:(1)取固相萃取柱(查标准是C18柱,但我印象中用的是HLB柱),安装在萃取装置上,关闭放液开关;(2)取烧杯,分别倒入少量甲醇、磷酸盐缓冲液、水以及流动相;(3)取吸管,用少量甲醇润洗3次,然后吸取甲醇2mL,加入固相柱,调节放液开关放液;(4)接近放完时,加入磷酸盐缓冲溶液2 mL,放液;(5)接近放完时,用单标线吸管加入上清液5.0 mL,放液;(6)放液完成后,加入水1mL淋洗,并用吸耳球吹干;(7)在装置下面放入接洗脱液的试管,放好装置后用流动相1.0 mL洗脱,挤干,收集洗脱液;(8)取1mL注射器去针头,吸取适量洗脱液,接上一次性滤器进行过滤,用进样瓶接取滤液,供上机测定。要点:(1)安装好固相柱后,一定要记得关闭放液开关;(2)几个烧杯要分别做好标记,以免混用;(3)活化柱子时要避免柱子干掉;(4)用一次性滤器过滤时,压力不宜过大,以免击穿滤膜。比赛至此,选手的现场操作基本结束。待次日检测数据及谱图出来后,填写原始记录并计算结果,上交赛事组织方即完成全部比赛内容。7比赛得失谈7.1失由于大多数选手是第一次参加此类大赛,很多问题都是预先没有考虑到的,还有一些问题是赛场压力造成发挥失常的,以下仅举几例:7.1.1吸管不润洗:少数选手拿到吸管,未经润洗直接吸液、放液。直到好几步操作下来后,发现旁边的选手(3个选手同时比赛相距不远)动作明显偏慢,才意识到自己的操作有问题。7.1.2离心前不平衡:有选手拿着2个平衡样,想直接放入离心机离心。结果被裁判直接叫停并扣分,那个选手还愣愣地不知道怎么回事,经裁判提醒后还以为2个待检样质量相同就不用平衡。7.1.3涡旋前未旋紧盖子。结果可想而知,回收率肯定不佳。7.1.4过柱时未充分发挥真空泵作用:活化时各位选手都选择自然放液,速度虽然有快有慢,但差异不显著,都还可以接受。但加入上清液后,绝大多数选手的放液速度都受到很大挑战,液体滴下的速度简直与蜗牛可以一拼。因为净化步骤有时间限制,有的选手就想着用吸耳球加压,结果效果不理想。看着时间只有不足2分钟,裁判老师们也急了,禁不住质问各位选手为何不使用真空泵?真是一语惊醒梦中人,各位选手马上启动真空泵,放液速度怎么还是这么慢?旁边的工作人员提醒各位选手,真空泵上有个真空度调节开关,顺时针旋就可以增加真空度。说得迟那时快,老师的话还没说完,几个选手几乎同时将真空度调节开关调到最大,只看到固相柱中的液体迅速排到下面试管中……7.2得7.2.1经历过比赛后,对标准的研读也会更加细致;7.2.2对操作的各个细节会更加注意,如做好标记、离心前先平衡、盖紧盖子再涡旋等等;7.2.3对仪器的操作做到提前了解细致,遇到未使用过的新仪器,比赛前要逐一先开机试用,掌握手感,做到比赛时心中有数;7.2.4最大的得当然还是比赛中拿到名次,比赛结束后半个多月,省农业厅等4部门联合发文,对优秀组织单位和个人予以通报表扬,本人有幸获得畜产品组三等奖。这是第一次参加如此高规格的比赛,说实在话除了以前参加过几次分析培训外,还真的没做过类似的实验,因此能有这个收获对自己来说还是挺有意义的。实际上这次比赛,最大的失与得,就在于学会了在净化过程中,应该合理利用固相萃取装置的负压,全程随时调节液体排放速度,做到既保持净化效果,又提高净化效率。







 我要推广仪器
我要推广仪器
 下载APP
下载APP