用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
六甲基二硅胺烷和三甲基氯硅烷作为硅甲基化试剂与醇的反应机理! 另外,六甲基二硅胺烷可以与醇反应,为什么还要加入三甲基氯硅烷?三甲基氯硅烷不是也可以与醇反应吗?请指教!
准备测定:乙醇分子中甲基和次甲基位点D/H含量的差异,请教,应该使用什么仪器,那个厂家的比较适合?谢谢!
群友问:用GB 5413.25-2010 第二法气相色谱法做肌醇,实验室买的三甲基氯硅烷和六甲基二硅胺烷有本底干扰,不知道老师们这两个试剂买的是什么牌子的?群友一:国药的群友二:三甲基氯硅烷,扫尾剂级别,100ml,国药有的,六甲基二硅胺烷,国扫尾剂级别,25ml,也是国药的;这两个试剂SIGMA有专用的衍生剂级别的。非常感谢乳制品检测之家群友岑老师、人气老师的分享!
请教各位大神,用啥柱子能把2-甲基丁醇和3-甲基丁醇在气相色谱图中分开呢?目前我用的是安捷伦 DB-WAX柱,柱子截过几次,两种醇的特征峰重叠了,无法区分。
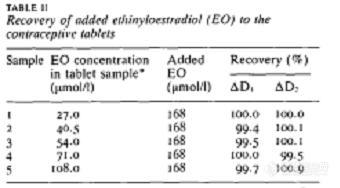
炔诺酮中炔雌醇的测定1 绪论采用各种已报道的方法测定避孕药片中乙炔雌二醇(EO)。这些方法包括分光光度法,高压液相色谱,薄层色谱和比色法。Corti等人用二阶导紫外分光测定二元混合物的雌激素和孕激素,相对标准偏差 5%以内。Ebeletal等人报告说,有几个方法确定EO的口服避孕药片,包括利用直接,差分 derivative紫外分光法和直角多项式法。作者报告说,后者的方法得到了最可靠的结果。最近, 事实已经证明,应用分光光度法的衍生技术是非常有益的,在解决光谱重叠, 消除来自其他样品不相干吸收。不过,在某些情况下,衍生的技术无法应付的干扰,尤其是当[size
最近进了个样品,固相微萃取进的样。有点脏。就用乙醇进了下空白针。结果发现,除了之前带的杂质外还有一个十二甲基环六硅氧烷峰。不知道是什么,有没有可能是柱流失?柱子是HP-5MS。
[color=#00008B][size=4]我们这边PVC生产中采用湿法制乙炔,对于乙炔纯度分析,采用的是丙酮吸收法。听说可以用二甲基甲酰胺做吸收剂,我试了一下,平常用丙酮吸收纯度分析结果为97.5%用二甲基甲酰胺做吸收剂有99.6%。请问乙炔纯度的分析方法那种吸收剂比较好?分析中哪些环节影响纯度分析?—————————————————————————————非常感谢![/size][/color]

测N-(甲氧甲基)-N-(三甲基硅甲基)苄胺我采用岛津[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],柱子为wax(聚乙二醇),所设参数SPL 290度,DFID 290度,柱温40度下保留1 min再以10度每分升温到240度,直接进样走到中间在某一峰后出现基线严重漂移,但基线可以回到零点,用甲醇稀释后基线漂移没有那么明显,而此处也显示有另外一个物质存在,想请教高手出现这种状况是什么原因?是物质沸点太高参数选择不对还是物质与柱子极性不匹配?N-(甲氧甲基)-N-(三甲基硅甲基)苄胺的沸点为76 º C (0.3 MMHG)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181274]N-(甲氧甲基)-N-(三甲基硅甲基)苄胺色谱图(甲醇溶解).doc[/url][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911051117_181361_1618994_3.jpg[/img]
分析二甲基丁醇,三甲基丁醇,样品浓度大约为99%。请教分析方法及使用何种色谱柱?谢谢了!![em58]
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
大家好: 一直以来 3-甲基-1-丁醇,3-甲基-2-丁醇的分离问题一直困扰着我,哪位高手能帮忙解决一下,请问用什么样的毛细柱分离,在什么条件下?先谢谢各位了。
RT?新购的气相色谱仪和柱子,只有上面两根柱子,一根是准备做中药材 醇类含量,一根是做农残的,省检定中心要来进行仪器检定,提前让我们准备好能做 正十六烷的 柱子,我想问下50%苯基甲基聚硅氧烷 能不能做?查阅资料,能做 烷烃的 有100%甲基聚硅氧烷(胶体和流体)、5%苯基聚硅氧烷, 我们现在有的PEG和 50%苯基甲基聚硅氧烷 能否做?万分感谢!
我用GC-ms, 柱是HP-35MS,打八甲基环四硅氧烷的时候发现,就是在溶剂中也会有很高的响应,不知道是什么原因?我用的溶剂是乙酸乙酯,发现溶剂 M/Z 281有很大的响应,最初怀疑是溶剂有问题,后来用LC的甲醇,发现也有同样的问题,说明不是溶剂的原因,大家有什么好的办解决吗?、
各位老师好: 现在做食品中的二甲级异茨醇,需要用微固相萃取技术,但是本实验室没有,请问时候可以用别的方法替代,请指点,谢谢!
求助:化妆品中的二甲基硅氧烷和混居二甲基硅氧烷如何提取啊?
各位大神,小弟最近在分析香精样品,目前用的DB-wax柱,但香精中的2-甲基丁醇和3-甲基丁醇峰分不开,请问各位大神用什么柱子在什么条件下可以使这俩种物质的峰完全分开呢?
领导让做聚丙烯酰胺的离子度,标准17514-2017里面提到甲基乙二醇甲壳素(MGC)标准溶液0.005mol/L,请问我要怎么采购,供应商那里说没有这东西,只有甲基乙二醇壳聚素
我在检验聚山梨酯80的环氧乙烷和二氧六环时,用的药典推荐聚二甲基硅氧烷色谱柱,没有峰,网上查看网友推荐是聚乙二醇色谱柱,大家都是怎么做的
买来的甲醇中甲基汞是69.5ug/g,若要配成中间储备液如何配?个人觉得是69.5*0.7918(甲醇密度)=55.03ppm 再进行稀释 但是身边有同事提出需要换算到汞的含量再稀释即 69.5/215.63(甲基汞摩尔质量)*201(汞摩尔质量)*0.7918(甲醇密度)=51.30ppm 现在思路比较乱,还请论坛上大神帮帮忙!万分感谢~
我用GC-FID测芝麻香型白酒中的3-甲基丙醇,为何检不到?请指点。
今天测试一个原材料 [i]2-甲基-1,3-丙二醇,发现多出一个峰,有没有分析的同行[/i]
1.烯烃、炔烃 、二烯能使溴的四氯化碳溶液,红色腿去,又能使高锰酸钾溶液,紫色腿去2.含有炔氢的炔烃(1)能使硝酸银,生成炔化银白色沉淀(2)又能使氯化亚铜的氨溶液,生成炔化亚铜红色沉淀。3.卤代烃:硝酸银的醇溶液,生成卤化银沉淀;不同结构的卤代烃生成沉淀的速度不同,叔卤代烃和烯丙式卤代烃最快,仲卤代烃次之,伯卤代烃需加热才出现沉淀。 4.小环烃:三、四元脂环烃可使溴的四氯化碳溶液腿色 5.醇:(1)与金属钠反应放出氢气(鉴别6个碳原子以下的醇);(2)用卢卡斯试剂鉴别伯、仲、叔醇,叔醇立刻变浑浊,仲醇放置后变浑浊,伯醇放置后也无变化。6.酚或烯醇类化合物:(1)用三氯化铁溶液产生颜色(苯酚产生兰紫色)。(2)苯酚与溴水生成三溴苯酚白色沉淀。7.羰基化合物:(1)鉴别所有的醛酮:2,4-二硝基苯肼,产生黄色或橙红色沉淀;(2)区别醛与酮用托伦试剂,醛能生成银镜,而酮不能;(3)区别芳香醛与脂肪醛或酮与脂肪醛,用斐林试剂,脂肪醛生成砖红色沉淀,而酮和芳香醛不能;(4)鉴别甲基酮和具有结构的醇,用碘的氢氧化钠溶液,生成黄色的碘仿沉淀。 8.甲酸:用托伦试剂,甲酸能生成银镜,而其他酸不能。 9.胺:区别伯、仲、叔胺有两种方法(1)用苯磺酰氯或对甲苯磺酰氯,在NaOH溶液中反应,伯胺生成的产物溶于NaOH;仲胺生成的产物不溶于NaOH溶液;叔胺不发生反应。(2)用NaNO2+HCl:脂肪胺:伯胺放出氮气,仲胺生成黄色油状物,叔胺不反应。芳香胺:伯胺生成重氮盐,仲胺生成黄色油状物,叔胺生成绿色固体。10.糖:(1)单糖都能与托伦试剂和斐林试剂作用,产生银镜或砖红色沉淀;(2)葡萄糖与果糖:用溴水可区别葡萄糖与果糖,葡萄糖能使溴水褪色,而果糖不能。(3)麦芽糖与蔗糖:用托伦试剂或斐林试剂,麦芽糖可生成银镜或砖红色沉淀,而蔗糖不能。
0.2%甲基红乙醇溶液中的甲基红不好溶解啊?我用0.2g甲基红+100ml乙醇,结果,甲基红很难溶解,该怎么配制?请高手告知,谢谢,

在甲基培尼皮质醇的制造过程中,可能会生成一些杂质。这些杂质可能会出现在原料中,也可能在制药过程中的化学反应中产生。不论其来源,杂质的存在都可能影响到药物的质量、安全性和疗效。例如,甲基培尼皮质杂质可能增加药物的毒性,或导致不良反应。同样,杂质也可能对甲基培尼皮质醇的药效产生影响。因此,制药公司必须在生产过程中严格检测和控制这些杂质。检测和控制药品中的杂质是药品质量控制的重要组成部分。CATO标准品对杂质的研究不仅有助于保证药品的质量和安全性,也可以为优化制药过程提供参考。比如,通过对杂质的研究,可以找到产生这些杂质的原因,从而改进制药过程,减少杂质的生成。[img=,600,588]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052058222428_1748_6381668_3.png!w600x588.jpg[/img]
大家好,本人菜鸟,(戴安ICS-90)想问下淋洗液甲基磺酸大家平时买哪些厂家的,纯度多少的?看了Sigma-Aldrich 纯度≥99.5% 价格有点贵,请各位不吝赐教。谢谢。
最近在扩关于生活饮用水中2-甲基异莰醇和土臭素,采用固相微萃取法对2-甲基异莰醇和土臭素进样萃取,土臭素的回收率在98-102%左右,很稳定但发现2-甲基异莰醇很不稳定,回收率也是不稳定,忽高忽低,想问一下大家有出现过以上情况吗?如何解决,有何建议?

用毛细管柱测定法莫替丁中的甲醇、乙醇和NN二甲基甲酰铵中的有机残留,氮气做载气,FID检测器;柱温初始温度40,保留3分钟,25度每分的升温速率到150度,保留5分钟。进样口温度200度,检测器250度。进样量2微升,手动进样。可是进针后有时候出现一个类似的溶剂峰,有时候又没有,而且同一个条件下进一个配制好的标准样品,出峰形状也不同,主要是后面的三个被测的有机溶剂峰形太难看,请大家帮忙分析一下,急死我了。传上来的图分别为:1图为不进样空走图,2、3图为公司自制超纯水(结果不同),4、5图为卖来的注射用水(也不一样),6、7图为自己配制的甲醇、乙醇、NN二甲基甲酰胺标准液(结果也不同)。以前验证过这个方法,就是这样的条件,就这三个峰,没有前面的那个类似溶剂峰的那个东西,现在不知道为什么时有时无那个峰,也出现过只出三个峰的时候,就是基线不太好,可是紧接着再进针峰形就变了。(对这个帖子进行了补充,以前写的有点不具体,谢谢大家的帮忙,我这几天一直关注这里,因为困扰我好几天了)[img]http://ng1.17img.cn/bbsfiles/images/2009/01/200901162150_129422_1614350_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/01/200901162151_129423_1614350_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/01/200901162152_129424_1614350_3.jpg[/img]
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
《六甲基二硅胺烷》的标准,谢谢先!!!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP