哪位朋友手头有GB/T 23967-2009 工业用偏苯三酸酐,可否上传上来啊!
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法
据说欧盟委员会在官方公报上发布决议(EU) 2018/594,正式将苯-1,2,4-三羧酸-1,2-酐(偏苯三酸酐)(TMA)确定为高度关注物质(SVHCs)。而在官网链接中还是181项[url]https://echa.europa.eu/web/guest/candidate-list-table[/url];是不是还没加进清单呢?[img]http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif[/img]
[color=#444444]偏苯三甲酸酐加氢产物分析,我做的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],液相分不开,质谱有一种物质没有离子化,没有峰,用的ESI源,请问用哪种色谱柱可以分开,怎样可以使样品离子化,谢谢[/color]
做实验要用到苯二甲酸酐,但是都买不到,都是只有邻苯二甲酸酐,问了几家试剂公司,有的说是一个东西,有的说不是,我在网上查苯二甲酸酐,出来的也都是邻苯二甲酸酐,到现在也没有确切的答案,但是国标方法上就写的苯二甲酸酐.
如何用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中分离邻苯二甲酸和邻苯二甲酸酐,或者怎么区分苯甲酸中存在的是邻苯二甲酸还是邻苯二甲酸酐,邻苯二甲酸在208度的时候会脱水转变为邻苯二甲酸酐。
国标上写的 苯二甲酸酐C6H40 邻苯二甲酸酐的化学式是 C8H4O3 是怎么回事呢,着急呀
收到两个样品,疑似为醋酸酐和三氯甲烷,待鉴定。我们实验室有agilent 气象色谱仪(配HP-5色谱柱)和气质联用仪(HP-5 MS柱和单四级杆)。据说质谱图图库里没有醋酸酐?不知道我们的条件是否可做改鉴定?应该如何检测?请高手指教,谢谢!
收到两个样品,疑似为醋酸酐和三氯甲烷,待鉴定。我们实验室有agilent 气象色谱仪(配HP-5色谱柱)和气质联用仪(HP-5 MS柱和单四级杆)。据说质谱图图库里没有醋酸酐?不知道我们的条件是否可做改鉴定?应该如何检测?请高手指教,谢谢!
SVHC第八批物质种有一组甲基六氢邻苯二甲酸酐,是由4个同分异构体组成,CAS号分别为:25550-51-0 19438-60-9 48122-14-1 57110-29-9但是就其分子式来看应该只有三种同分异构体,为什么会有4个CAS号呢,或者是船式和椅式的构象不同造成的4种同分异构体?
收到两个样品,疑似为醋酸酐和三氯甲烷,待鉴定。我们实验室有agilent 气象色谱仪(配HP-5色谱柱)和气质联用仪(HP-5 MS柱和单四级杆)。据说质谱图图库里没有醋酸酐?不知道我们的条件是否可做改鉴定?应该如何检测?请高手指教,谢谢!
一个实例,用七氟丁酸酐可以与二苯基甲烷二胺发生衍生化反应,衍生化的机理应该在于胺根上面。但二苯基甲烷二胺里面的胺还会与羧酸发生反应。如果同时向含有 大量的七氟丁酸酐 和 极少量的 丁酸 的混合溶剂中,加入少量二苯基甲烷二胺,请问二苯基甲烷二胺是先发生衍生化反应还是与丁酸反应?
工作场所中邻苯二甲酸酐的测定 依据GBZ/T160.60-2004 用的毛细管柱DB-WAX 只有溶剂丙酮的峰 邻苯二甲酸酐没岀峰 是柱子不合适吗?
我按GBZ/T160.60-2004的标准,改用毛细管色谱柱分析邻苯二甲酸酐时,怎么也不出峰,有做过的大侠指点下。
做水,乙酸,甲苯,马来酸酐,糠醛,苯甲醛,丙烯醛用什么色谱柱!
求助三氟甲磺酸酐气相测试方法我们用HP-5测试 不知道测试出来的是不是主峰,并且杂质较多,求助测试方法?是否可以做硅化测试?
我所用的仪器是安捷伦6820 柱子DB-17 用什么方法能查出对苯二甲酸酐里含有邻苯二甲酸吗
4‘4-氧双邻苯二甲酸酐 cas 1823-59-2,苯醚相对稳定,但酸酐遇水容易水解成酸:常温常压下反应比较慢,加热和遇到酸碱会加快反应。请问各位大侠有没有好的检测方法?

今天在研读农业部781号公告-5-2006 《动物源食品中阿维菌素类药物残留量的测定 高效液相色谱法》,发现里面关于三氟乙酸酐的稀释有问题:1.在4.2试剂和材料中写的稀释比例是1:2http://ng1.17img.cn/bbsfiles/images/2016/01/201601271633_583704_1627156_3.jpg2.在4.4.3净化和荧光衍生化以及4.4.4标准曲线的测定中写的稀释比例是1:1http://ng1.17img.cn/bbsfiles/images/2016/01/201601271633_583705_1627156_3.jpg前后两段的描述不一致,究竟稀释比例是1:1还是1:2?3.查阅其它标准,GB29696-2013 《食品安全国家标准 牛奶中阿维菌素类药物多残留的测定 高效液相色谱法》中有如下描述:http://ng1.17img.cn/bbsfiles/images/2016/01/201601271638_583711_1627156_3.jpg参考GB29696-2013的描述,个人认为三氟乙酸酐稀释比例应该为1:2,农业部781号公告-5-2006的4.4.3和4.4.4的稀释比例1:1应该笔误。诸位大大意下如何,不妨讨论一下
根据GBZ/T 160.67-2004 扩项工作场所MDI。原理:空气中MDI用冲击式吸收管采集,水解后成4.4’-二氨基二苯甲烷(MDA),在碱性条件下用甲苯萃取,经七氟丁酸酐衍生后,取甲苯溶液进样,经色谱柱分离,电子捕获检测器检测,以保留时间定性,峰面积定量。标准曲线的绘制:在5 只干燥的具塞离心管中,0.0、0.25、0.50、1.0和2.0ml MDA标准溶液,用甲苯稀释至2.0ml,配制成0.0、0.025、0.050、0.10和0.20mg/ml MDA标准系列,各管加30ul 七氟丁酸酐,振摇2min,放置5min,加1ml 缓冲液,振摇2min,以除去过剩的七氟丁酸酐,放置2min,将甲苯层转移入另一离心管中,供测定。色谱柱 DB-5 柱温230℃ 进样器270℃,检测器250℃,结果只出甲苯溶剂峰。参考了些文献七氟丁酸酐与胺反应有加热55℃70分钟的,也有反应30min的。标准里衍生反应很短时间怀疑衍生反应有问题!
各位专家好! 现需求三氟乙酸酐国家标准,请大家多帮忙! 如果有的话,请提供资料,没有的话,也请大家提供一下检测方法。 谢谢!
问一下 正在做邻苯二甲酸酐,按的是GBZ/T160.60-2004的方法,色谱条件是柱温170 汽化室200 检测室200 用FFAP填充柱做的 只有一个丙酮的平头峰,该怎么做呢
三氟乙酸酐会腐蚀气相色谱系统吗, 7890-FID,可以用气相分析吗,谢谢大家啦
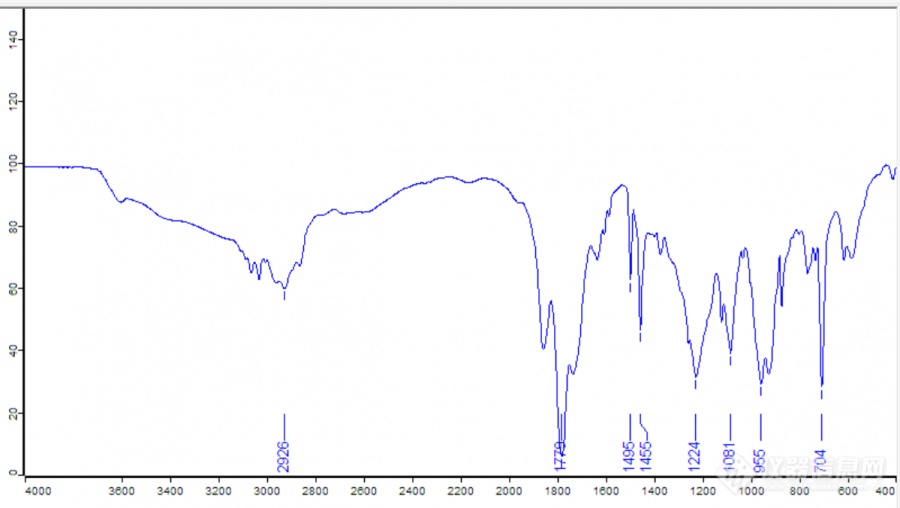
[table=100%][tr][td]苯乙烯-马来酸酐的红外光谱图跟文献好像差不多,怎么看是否水解?光谱图见附件。GPC居然没有测出来,没有任何峰,GPC测试老师说可能是聚合物被吸附在了柱子的填料上了?? GPC图见附件。我是把反应后的聚合物用正己烷沉淀出来,然后放入60度烘箱中干燥,再取出溶于THF中,拿去测试中心测试。文献中也是溶于四氢呋喃啊,没有看到有什么特殊的处理啊。如果没有水解,酸酐也容易吸附在GPC柱子上吗??[/td][/tr][/table][img=,690,389]https://ng1.17img.cn/bbsfiles/images/2019/08/201908301048373430_6450_1801607_3.png!w690x389.jpg[/img][img=,690,304]https://ng1.17img.cn/bbsfiles/images/2019/08/201908301048386830_8910_1801607_3.png!w690x304.jpg[/img]
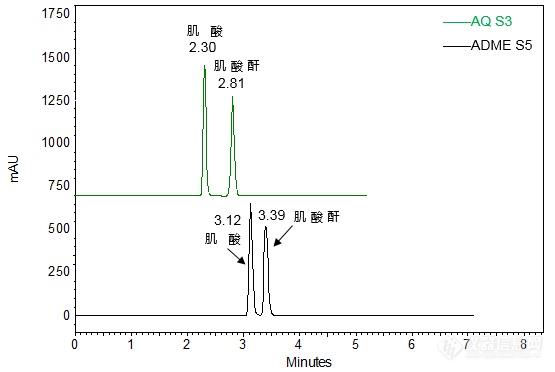
[align=center][b]肌酸和肌酸酐的分析[/b][/align][b][/b]客户提供肌酸、肌酸酐样品,希望实验室帮忙实现二者的良好分离与保留。由于肌酸和肌酸酐极性较强,我们首先尝试在酸性条件下(0.05%磷酸),使用高表面极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S3与CAPCELL PAK ADME S5两款色谱柱,分别对二者混合样品进行分析,结果如图1所示,肌酸的保留时间分别为2.30 和3.12 min,保留较弱。[align=center][img=,555,372]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021020_01_2222981_3.png!w555x372.jpg[/img][/align][align=center]图1 肌酸、肌酸酐混合溶液分析色谱图(0.05%磷酸溶液)[/align]*注:峰上标数字为保留时间。[img=,419,152]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021020_02_2222981_3.png!w419x152.jpg[/img]为增强保留,尝试在中性条件下,以200 mmol/L高氯酸钠溶液为流动相对肌酸和肌酸酐进行分析,结果如图2所示。使用AQ S3色谱柱进行分析时,肌酸、肌酸酐的保留时间分别为3.36 min、5.23min。使用ADME S5色谱柱进行分析时,肌酸、肌酸酐的保留时间分别为3.48 min、6.29 min。ADME 色谱柱较AQ S3 色谱柱整体保留较强。[align=center][img=,550,378]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021022_01_2222981_3.png!w550x378.jpg[/img][/align][align=center]图2 肌酸、肌酸酐混合溶液分析色谱图(200mmol/L高氯酸钠溶液)[/align]*注:峰上标数字为保留时间。[img=,420,149]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021022_02_2222981_3.png!w420x149.jpg[/img]进一步将200mmol/L高氯酸钠溶液的pH调至酸性(pH=2.74),以ADME S5色谱柱对肌酸、肌酸酐混合样品进行分析,结果如图3所示。肌酸、肌酸酐保留时间分别延长至4.45、5.07,二者间分离度为3.12,达到基线分离。[align=center][img=,552,372]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021023_01_2222981_3.png!w552x372.jpg[/img][/align][align=center]图3 肌酸、肌酸酐混合溶液分析色谱图(200 mmol/L高氯酸钠溶液、pH 2.74)[/align]*注:峰上标数字由下至上依次为分离度与保留时间。[img=,407,138]http://ng1.17img.cn/bbsfiles/images/2017/11/201711021023_02_2222981_3.png!w407x138.jpg[/img]
如何将样品中醋酸和醋酸酐的峰分开
[color=#444444]我用的是安捷伦7820[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]分析丝氨醇,三氟乙酸酐衍生,以前一直很正常,单杂都是小于0.1%,但是近来在主峰后出现一个0.2%左右的杂质,与异丝氨醇相近,客户没有检出!邮寄至其他研究所也没有检出!可是我衬管更换了,进样隔垫也更换了,色谱柱也更换了,氮气也更换了,进样针也更换了,都没有用,并且改杂质忽大忽小!空白都没有,衍生试剂三氟乙酸酐也更换了都没有用,请各位大神帮忙分析一下,谢谢啦!!![/color]
有哪些高人做过1、糠醇 2、顺丁烯二酸酐 3、甲基丙烯酸甘油酯 4、苯磺酸 5、胺菊酯、氯菊酯 。它们的GC条件是什么啊?急等回音,谢谢!
五氯苯酚中的试剂乙酸酐大家都用什么品牌,这个现在还在限制使用中吗?最近准备开展这个。
如何用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法分析乙酸和乙酸酐?







 我要推广仪器
我要推广仪器
 下载APP
下载APP