一阶导数分光光度法测定盐酸普鲁卡因溶液的含量刘素琴(江苏省金坛市人民医院,江苏 金坛 213200)联系电话:0519-2266680 E-mail:Liusuqin666@163.com 文章编号:04040399摘要 目的:改进盐酸普鲁卡因溶液的含量测定方法。 方法:以一阶导数光谱在308.0nm波长处谷—零间的振幅为定量依据,测定盐酸普鲁卡因溶液的含量。结果:盐酸普鲁卡因溶液浓度在5~30μg/mL范围内与一阶导数谷—零间的振幅呈良好的线性关系,r=0.9999,平均回收率为100.04%,RSD=0.43%。结论:方法简便、准确,可用于测定盐酸普鲁卡因溶液的含量。关键词 一阶导数; 分光光度法; 盐酸普鲁卡因 ;含量[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=32338]一阶导数分光光度法测定盐酸普鲁卡因溶液的含量[/url]
第一章 麻醉药 第一节 全身麻醉药 1、 吸入麻醉药 氟烷:2-溴-2-氯-1,1,1-三氟乙烷 起效、苏醒快、作用弱,全麻及诱导麻醉 性质:1、氧瓶燃烧 2、加入硫酸,沉于底部。 甲氧氟烷浮于硫酸上层。 甲氧氟烷:麻醉作用和肌松作用比氟烷强,诱导期长。 恩氟烷:新型高效吸入麻醉药,麻醉肌松作用强,起效快,临床常用。 异氟烷为异构体 乙醚:氧化后生成过氧化物对呼吸道有刺激作用。 2、 静脉麻醉药 盐酸氯胺酮:2-(2-氯苯基)-2-(甲氨基)环已酮盐酸盐 2个旋光异构体,用外消旋体 作用快、短、副作用小,诱导期短。 分离麻醉 羟丁酸钠: 作用弱、慢、毒性小。 --OH 1、三氯化铁红色 2、硝酸铈铵橙红色 第二节 局部麻醉药 一、 对氨基苯甲酸酯类 构效关系:1、苯环上增加共他取代基时,因增加空间位阻酯基水解减慢,局麻作用增强。 2、苯环上氨基的烃以烷基取代,增强局麻作用。 丁卡因 3、改变侧链氨基的取代基,有些作用增强。 布他卡因 4、羧酸中的氧原子若以电子等排体硫原子替代(硫卡因),脂溶性增大,作用增强。 盐酸普鲁卡因:4-氨基苯甲酸-2-(二乙氨基)乙酯盐酸盐 不宜表面麻醉 性质:1、加氢氧化钠有油状普鲁卡因析出。 干燥稳定,避光 PH=3-3.5最稳定。 2、酯键:水溶液水解失活:对氨基苯甲酸及二乙氨基乙醇,前者氧化变色 3、叔胺结构:碘、苦味酸等呈色 4、芳伯氨反应: 盐酸丁卡因:4-(丁氨基)苯甲酸-2-(二甲氨基)乙酯盐酸盐 作用:用于粘膜麻醉,与普鲁卡因一起成为应用最广的局麻药。 二、酰胺类: 盐酸利多卡因:N-(2,6-二甲基苯基)-2-(二乙氨基)-乙酰胺盐酸盐-水合物 性质:酰胺键较酯键稳定,酸碱中均较稳定 。作用强,可用于表面麻醉 布比卡因:1-丁基-N-(2,6-二甲苯基)-2-哌啶甲酰胺盐酸盐 长效局麻药,用于浸润麻醉。 三、氨基酮类及氨基醚类
测定药品(丙胺卡因,利多卡因)中二氯甲烷和N,N-二甲基甲酰胺
第十章 胺类药物的分析掌握盐酸普鲁卡因、盐酸利多卡因、盐酸丁卡因和对乙酰氨基酚的鉴别、杂质检查和含量测定方法。 掌握肾上腺素、盐酸去氧肾上腺素及其制剂的鉴别、杂质检查和含量测定方法。 第一节 盐酸普鲁卡因的分析有芳伯氨基特性,显重氮化-偶合反应。含酯键易水解,产物主要为对氨基苯甲酸(PABA)。脂烃胺侧链为叔胺氮原子,具有弱碱性。盐酸盐系白色结晶性粉末,具有一定的熔点,易溶于水和乙醇,难溶于有机溶剂。一、鉴别1.重氮化-偶合反应 分子结构中具有芳伯氨基或潜在芳伯氨基的药物,均可发生重氮化反应,生成的重氮盐可与碱性β-萘酚偶合生成有色的偶氮染料。 2.水解反应 取本品约0.1g,加水2ml溶解后,加10%氢氧化钠溶液1ml,即生成白色沉淀,加热变为油状物(普鲁卡因);继续加热,产生的蒸汽(二乙氨基乙醇)能使湿润的红色石蕊试纸变为蓝色;热至油状物消失后(生成可溶于水的对氨基苯甲酸钠),放冷,加酸酸化,即析出白色沉淀。此沉淀能溶于过量的盐酸。3.氯化物反应沉淀反应 在硝酸酸性条件下与硝酸银生成白色沉淀,沉淀加氨试液即溶解。氧化还原反应 加二氧化锰混匀,硫酸润湿,加热产生氯气,能使湿润淀粉碘化钾试纸显蓝色。4.红外光谱法二、特殊杂质检查普鲁卡因分子结构中有酯键,易发生水解反应。其注射液制备过程中受灭菌温度、时间、溶液pH值、贮藏时间以及光线和金属离子等因素的影响,可发生水解反应生成对氨基苯甲酸和二乙氨基醇。其中对氨基苯甲酸随贮藏时间的延长或高温加热,可进一步脱羧转化为苯胺,而苯胺又可被氧化为有色物,使注射液变黄,疗效下降,毒性增加。故中国药典90年版规定,本品注射液应检查水解产物对氨基苯甲酸,其限度不得超过1.2%。检查方法 薄层色谱法。供试品溶液2.5mg/ml与对照品溶液30μg/ml分别点于硅胶H薄层板上,展开后用对二甲氨基苯甲醛溶液喷雾显色。供试品溶液如显与对照品溶液相应的杂质斑点,其颜色与对照品溶液主斑点比较,不得更深。三、含量测定 分子结构中具有芳伯氨基,可用亚硝酸钠滴定法测定含量。
今天发现有些原料药没有用红外鉴别,比如胺类药物:普鲁卡因、利多卡因、对乙酰氨基酚、肾上腺素中,肾上腺素没有使用红外鉴别,为什么?IR的鉴别力不是很强吗?为什么不收入药典?苯巴比妥类药物,苯巴比妥、司可巴比妥钠、硫喷妥钠中,硫喷妥钠没有使用IR鉴别,why?
[color=#DC143C][B]下面有附件请点击保存[/B][/color]h1,h2 相邻谱峰1和谱峰2的峰高IEC 离子交换色谱法 Ion-exchange chromatography IP 离子对 Ion-pair IPC 离子对色谱法 Ion-pair chromatography J 色谱峰强度参数K’所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’) k 梯度洗脱过程中,某溶质的k’的平均值或有效值kw 以水做流动相k’的外推值k1,k2 相邻谱峰1和谱峰2的容量因子L 色谱柱长度(cm)Lc 检测器流动池光路的长度(cm)M 溶质的分子量MC 二氯甲烷 Methylene chloride MDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件MeOH 甲醇 Methanol MTBE 甲基叔丁醚 Methyl-t-butyl ether MW 溶质的分子量N 色谱柱塔板数NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质)N0 检测器的基线噪音ODS 十八烷基硅烷 Octadecylsilyl P 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数PA 普鲁卡因胺 Procainamide(碱性物质)PAH 聚芳香烃 Polyaromatic Hydrocarbon PESOS 优化流动相的计算机软件(美国Perkin-Elmer产品)pKa 溶质酸性常数的负对数;当pH=pKa时,溶质中有一半是电离的Rk 保留值范围,Rk=(最末谱峰k’)/(最初谱峰k’)RRM 相对分离度图(通常N=10000)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=67452]【分享】色谱缩略语[/url]

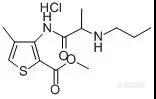
作者:李昂; 胡翮; 郭丙炎;(湖南省湘潭市药品检验所;)摘要:目的用反相高效液相色谱(RP-HPLC)法测定盐酸丁卡因的含量。方法色谱柱为Diamonsil C18柱(150mm×4.6mm,5μm),流动相为水-甲醇-乙腈(45∶45∶10,v/v,含0.02%庚烷磺酸钠,0.34%磷酸二氢钾,三乙胺调pH至7.0),流速为1.0mL/min,检测波长为314nm,柱温为室温。结果盐酸丁卡因质量浓度在5.034~161.1μg/mL范围内与峰面积线性关系良好,回归方程为Y=5.6572×105X-9.2039×105(r=0.9999),平均回收率为100.76%,RSD为0.41%(n=6)。结论RP-HPLC法简便、可靠、准确,可作为盐酸丁卡因注射液的含量测定方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271103_386360_1606903_3.jpg
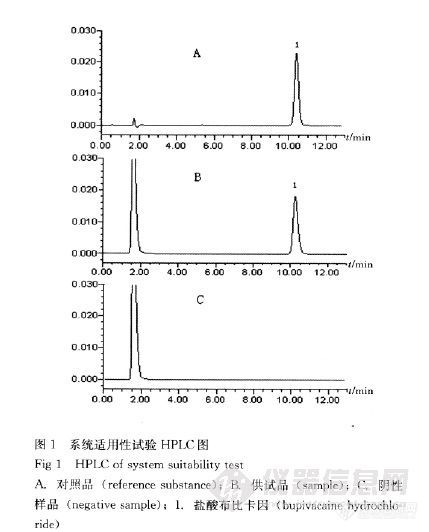
在盐酸普鲁卡因检查项中,用高效液相色谱法测定对氨基苯甲酸的限度,在系统适用性实验中,为什么将供试品溶液和对照品溶液按照一定比例混合之后进样呢?
青霉素治畜禽病须注意1.青霉素不能与碱性药物配合。在兽医临床中,常见的属于此种错误的配伍,为青霉素与碘胺类钠盐注射液或与碳酸氢钠注射液配伍。 2.青霉素不能与酸性药物配伍。有人把青霉素与土霉素、链霉素一起用蒸馏水溶解,给雏鸡喷雾,治疗传染性支气管炎。这是不对的。因土霉素溶液ph值为2~2.9,属于酸性较强的药物。ph值5以下的酸性药物还有很多,如四环素注射液、肾上腺素注射液、盐酸山梗菜碱注射液、注射用三磷酸腺苷、葡萄糖注射液、氯化钾注射液、氯化钙注射液、山梨醇注射液、甘露醇注射液、注射用促皮质素、杜冷丁注射液、盐酸氯丙嗪注射液、脑垂体后叶注射液、马来酸麦角新碱注射、催产素注射液等,都不可与青霉素配伍。 3.青霉素不能与氢化可的松注射液配伍。2%以下的醇,对青霉素无破坏作用;高于2%时,则有轻微破坏作用;25%以上的醇,可使青霉素失效。 4.青霉素不能与配伍后发生浑浊、沉淀和降低效价的注射剂配伍。如硫酸卡那霉素注射液、注射用辅酶a、注射用细胞色素c、氨茶碱注射液、盐酸异丙嗪注射液、注射用乳糖酸红霉素、注射用硫喷妥钠等。 5.青霉素不能与高浓度的盐酸普鲁卡因溶液配伍。盐酸普鲁卡因的最适宜浓度为0.25%~0.5%。浓度过高,则起相反作用。 6.单胃动物如猪、驴、骡、马,不能口服青霉素。多胃动物如牛和羊,可以口服青霉素。 7.青霉素必须重复使用。否则,那些漏网的和新繁殖的微生物,会产生抗药性,危害更烈。必须每隔4~6小时重复用药1次。用有延缓青霉素在体内作用时间的溶媒(如0.25%~0.5%的盐酸普鲁卡因)溶解青霉素,可每隔8~12小时重复用药1次。 8.家畜发生青霉素过敏反应的抢救。兽医界无作过敏试验的规定,用药前先询问畜主患畜有没有青霉素过敏史,如有,就改用其他抗菌素;如无,在注射青霉素后,要至少观察半小时,无反应时,方视作安全。家畜发生青霉素过敏,应立即抢救。可用0.1%盐酸肾上腺素注射液,皮下注射。用量:猪、羊、驹、犊0.2~1毫升,牛、马5毫升。也可稀释10倍静注,用量减半。也可静注10%葡萄糖酸钙注射液,猪、羊、驹、犊200毫升,牛、马500毫升。

10,抽取5个版友);中奖名单:玲儿响叮当(注册ID:jshbhh)捌道巴拉巴巴巴(注册ID:v3082413)大川之子,纵横四海(注册ID:chuangu120)翠湖园(注册ID:hhx050)sixingxing(注册ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/09/201609231507_611891_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/09/201609231507_611892_1610895_3.png【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================血浆中利多卡因的测定方法:SPE基质:血浆应用编号:101288化合物:利多卡因固定相:ProElut PLS色谱柱/前处理小柱:ProElut PLS 30mg / 1ml 100/pkg样品前处理:1、样品准备 制备2 - 3 mL 血浆,取1 mL 血浆与1 mL 水混匀,待净化。2、净化 a 活化: 依次将1 mL 甲醇、1 mL 水加入ProElut PLS 30 mg/1 mL (Cat.#68002),流出液弃去; b 上样: 将样品加入小柱,流出液弃去; c 淋洗: 1 mL 5% 甲醇水溶液淋洗,淋洗液弃去,将小柱抽干; d 洗脱: 1 mL 甲醇洗脱,收集洗脱液; e 重新溶解:40 oC 下氮气吹干洗脱液,1 mL 流动相定容色谱条件:色谱柱:Diamonsil C18(2) 200 x 4.6 mm ID, 5 μm (Cat. #99602) HPLC应用101350文章出处:P040关键字:血浆,利多卡因,SPE,ProElut PLS摘要:适用于人与动物血浆中利多卡因的检测。谱图:http://www.dikma.com.cn/Public/Uploads/images/p91%20copy.png图例:1. 立多卡因
液相色谱常用符号与术语表『转贴』ACN 乙腈 Acetonitrile AUFS 满量程的吸光度单位 Absorbance units, full scale As 峰不对称因子 B 二元流动相中的强溶剂;例如:反相HPLC的甲醇/水混合液中的甲醇 BSA 牛血清白蛋白(一种蛋白质) Bovine serum albumin CAF 咖啡因(中性溶质) Caffeine CRF 色谱响应因子 Chromatographic response function;色谱图总分离度的定量指标 dc 色谱柱内径(cm) DMOA 二甲基辛胺 Dimethyloctylamine DNB 2,4-二硝基甲酰(基) 2,4-Dinitrobenzoyl dp 色谱柱填料的粒度(cm) DRYLAB 液相资源公司(LC Resources INC.)的计算机模拟软件。DRYLAB I用于等度预测,DRYLAB G用于梯度预测 F 流动相的流速(ml/min) FC-113 1,1,2-三氟-1,2,2-三氯乙烷 GPC 凝胶渗透色谱法 Gel-permeation chromatography HA 酸性溶质,能电离出A- Hex 己烷 Hexane hr 二相邻谱带之间的谷高 HVA 高香草酸 Homovanillic acid h’ 峰高 h1,h2 相邻谱峰1和谱峰2的峰高 IEC 离子交换色谱法 Ion-exchange chromatography IP 离子对 Ion-pair IPC 离子对色谱法 Ion-pair chromatography J 色谱峰强度参数 K’ 所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’) k 梯度洗脱过程中,某溶质的k’的平均值或有效值 kw 以水做流动相k’的外推值 k1,k2 相邻谱峰1和谱峰2的容量因子 L 色谱柱长度(cm) Lc 检测器流动池光路的长度(cm) M 溶质的分子量 MC 二氯甲烷 Methylene chloride MDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件 MeOH 甲醇 Methanol MTBE 甲基叔丁醚 Methyl-t-butyl ether MW 溶质的分子量 N 色谱柱塔板数 NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质) N0 检测器的基线噪音 ODS 十八烷基硅烷 Octadecylsilyl P 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数 PA 普鲁卡因胺 Procainamide(碱性物质) PAH 聚芳香烃 Polyaromatic Hydrocarbon PESOS 优化流动相的计算机软件(美国Perkin-Elmer产品) pKa 溶质酸性常数的负对数;当pH=pKa时,溶质中有一半是电离的 Rk 保留值范围,Rk=(最末谱峰k’)/(最初谱峰k’) RRM 相对分离度图(通常N=10000) Rs 相邻二谱峰的分离度 S 当流动相中的%B改变时,测量溶质保留值的变化速率的参数 SAL 水杨酸 Salicylic Acid SEC 尺寸排阻色谱法 Size-exclusion chromatography S/N 信噪比 Signal to noise ratio t 分离时间(min)(样品进样时t=0) tp 梯度系统的滞后时间(min) TBA 四丁基铵离子 Tetrabutylammonium ion TEA 三乙胺 Triethylamine THF 四氢呋喃 Tetrahydrofuran tk 在用于校正等度洗脱溶剂强度的流动相离开梯度混合器时,梯度洗脱的时间 TLC 薄层色谱法 Thin-layer chromatography TMA 四甲基铵 Tetramethylammonium(盐) TMS 三甲基硅烷 Trimethylsilyl t0 色谱柱的死时间(min) tR 溶质的保留时间(min) tG 梯度时间(min),即梯度开始至结束的时间 t1,t2 相邻谱峰1和谱峰2的保留时间(min) ti 色谱图中第一峰的保留时间(min) tf 色谱图中最末峰的保留时间(min) △tg tf-ti tx (tf-ti)/2 UV 紫外光 Vm 色谱柱的死体积(mL),Vm=t0F VMA 香草扁桃酸 Vanillymandelic acid wm 化合物的进样量 w1,w2 相邻谱峰1和谱峰2于半峰高处(W1/2)的宽度(min) W1,W2 相邻谱峰1和谱峰2的基线宽度(min) W1/2 半峰高处的谱带宽度 xd,xe,xn 溶剂选择参数,分别用于测定溶剂的酸度、碱度和偶极性的程度 α 分离因子,α=k2/k1 △Φ 梯度洗脱期间流动相成分的变化 εo 溶剂强度参数 ε 化合物的克分子吸收系数 η 流动相的粘度(Pas) Φ 流动相中强溶剂的体积份数 %B 二元流动相中强溶剂的体积百分比(%v)[em09]
海洛因、咖啡因的FTIR检验及谱图解释 摘要: 目的提高用红外光谱法鉴定海洛因、咖啡因纯品、混合物伪品的水平。方法用红外光谱法有针对性地选择特征峰,探明海洛因、咖啡因红外光谱与结构的关系。结果获得海洛因、咖啡因的特征峰。结论该方法克服了鉴定中的盲目性。 关键词:海洛因;咖啡因;红外光谱 Analysis of heroin and caffeine by FTIR and interpretations of the IR spectra FENGji-min ,LIU Shi-hai ABSTRACT: Objective To enhanced the capability to identify and differentiate the pure/mixture as well as some fakes heroin and caffeine.Method Explain away relationship between the spectra with structure characteristics of heroin and caffeine.Result Characteristic peaks.for beroin and caffeine were selected、Conclusion Pure/mixture as well as some fakes heroin and caffeine can be identified accurately with FTIR. KEY WORDS: heroin;caffeine;infrared spectroscopy 红外光谱法是国际上常用的毒品检验方法。红外光谱分析毒品主要有以下几个特点:所需检材量小,一般只需微克级;不破坏检材,样品仍可进行其它分析;操作简便,不需要特殊的前处理,10分钟可以完成一个样品分析;不需要其它试剂,不污染环境,有益于操作人员健康;重现性好,特异性强;适应性广,受杂质种类限制少。 目前有关红外光谱检验毒品的文献,大都只介绍了各种毒品纯品的红外光谱,很少对其吸收带做必要的解释。而不了解各吸收带与结构的对应关系做毒品鉴定,带有极大的盲目性,特别是分析混有杂质的毒品,无论是计算机检索,还是人工识图,都十分困难。鉴于目前这种情况,笔者在大量实验的基础上对海洛因盐酸盐和咖啡因的红外光谱解释做了初步尝试,以期与国内同行进行交流。 1 海洛因纯品的红外光谱及其解释 海洛因(heroin),是吗啡的衍生物,其盐酸盐的化学结构式见图。 海洛因盐酸盐的红外光谱中3439/cm为N-H的伸缩振动吸收,2957/cm为3个CH3中C-H反对称伸缩振动的偶合,2632、2524、2083/cm 是海洛因盐酸盐中叔胺盐离子的N+-H对称和反对称伸缩振动吸收。1762/cm为与苯环相连的乙酰氧基的羰基的伸缩振动,1738/cm为与6元环相连的乙酰氧基的羰基的伸缩振动吸收。二个羰基的伸缩振动之所以峰位有差别,是因为苯环的电负性比6元环的吸电性强,屏蔽作用大。吸电子的诱导效应使成键的电子密度向键的几何中心接近,降低了羰基C=0的极性,增加了双键性,伸缩振动力常数增加。吸电子基团的诱导效应和苯环上大的取代基的屏蔽作用导致羰基伸缩振动更多的向高频位移。1242和1037/cm两条谱带分别归属于=C-O-C=0中O-C=0和-O-C=的反对称和对称伸缩振动,是乙酰氧基光谱中两条最特征的谱带。两者和1761、1737/cm谱带结合起来指示酯类的存在。1630/cm为六元环上C=C的伸缩振动,乙酰氧基的强吸电性使其强度增大。1492、1445/cm是苯环的骨架振动。1445/cm也包含=N-CH3中C-H的变形振动吸收。 1469、1369/cm谱带分别为甲基的反对称变角振动和对称变角振动。它们的强度明显增强,是由于甲基直接和羰基相连所引起的,是乙酰结构的明显特征。1180、1157、1131为C-O的反对称伸缩振动和对称伸缩振动。1106/cm为同时与苯环和6元环相连的C-O-C键的伸缩振动。911/cm属于六元环上-CH=CH-中反式CH面外弯曲振动。764、696/cm为苯环上2个相邻氢原子的面外变角振动吸收。 2 咖啡因纯品的红外光谱及其解释咖啡因(caffeine)又称咖啡碱。由茶叶或咖啡中提得的一种生物碱。也可人工合成。呈白色晶体或粉末。密度1.23。熔点234-237℃。小剂量作用于大脑皮层高位部分,促使精神兴奋,较大剂量可直接兴奋呼吸中枢和血管运动中枢。1996年1月国家卫生部将其列入第一类精神药品。 1695/cm是2位碳原子上羰基C=O的伸缩振动,由于受邻位N原子诱导效应的影响所以出现在较高的波数。1659/cm是6位碳原子上C=O的伸缩振动,它既受邻位N原子诱导效应的影响,也与邻位4、5原子间的双键形成共轭。共轭使C=O双键特性减弱,力常数降低,伸缩振动向低频位移,同时强度增加。3114/cm是芳香环C-H的伸缩振动,其变角振动出现在745/cm。4、5原子C=C伸缩振动和8、9原子C=N伸缩振动的叠加出现在1600/cm和1551/cm。N-CH3的C-H伸缩振动出现在2959/cm,CH3不仅受N原子的影响,而且N原子也受邻近原子的影响。3个N-CH3的N原子与不同的原子相连,C-H变角振动出现在多个位置,它们分别是1485、1456、1426、1404/cmo O=C-N 中C-N的伸缩振动分别出现在1551和1360/cm。N3-C4的伸缩振动出现在1026/cm。1074/cm为C5-C6的伸缩振动。 3 海洛因和咖啡因混合物的检验 从毒贩手中缴获的海洛因等毒品中经常掺杂有咖啡因。例如从云南发生的一起贩毒大案中缴获的毒品(公安部物证鉴定中心编为滇111号),经红外光谱检验,其红外光谱如图。把它与前面两图相比较可以发现:3116、2957、1705、1660、1549、1284、1241、974、761、745、611、480、447、426/cm为咖啡因的红外吸收;3437、2957、2634、2088、1762、1737、1489、1445 1366 1241 1182 1156 1132、1109 1033、974、912、873、837、798、761、698、644、522/cm为海洛因盐酸盐的红外吸收。经进一步检验得知,该样品中海洛因盐酸盐与咖啡因的质量比为1:0.8。 样品2则是上海一起贩毒大案中缴获的毒品(公安部物证鉴定中心编为沪85号),经红外光谱检验,其红外光谱图与前两图相比较可以发现 3113、2955、1696、1658、1549、1449、1287、1241975、748、446、423/cm为咖啡因的红外吸收。3446、2955 2634 2086 1763 1735 1487、1241 1106、1037、975、910、873、798、696、571、472/cm为海洛因盐酸盐的红外吸收。
液相色谱常用符号与术语表ACN 乙腈 Acetonitrile AUFS 满量程的吸光度单位 Absorbance units, full scale As 峰不对称因子 B 二元流动相中的强溶剂;例如:反相HPLC的甲醇/水混合液中的甲醇 BSA 牛血清白蛋白(一种蛋白质) Bovine serum albumin CAF 咖啡因(中性溶质) Caffeine CRF 色谱响应因子 Chromatographic response function;色谱图总分离度的定量指标 dc 色谱柱内径(cm) DMOA 二甲基辛胺 Dimethyloctylamine DNB 2,4-二硝基甲酰(基) 2,4-Dinitrobenzoyl dp 色谱柱填料的粒度(cm) DRYLAB 液相资源公司(LC Resources INC.)的计算机模拟软件。DRYLAB I用于等度预测,DRYLAB G用于梯度预测 F 流动相的流速(ml/min) FC-113 1,1,2-三氟-1,2,2-三氯乙烷 GPC 凝胶渗透色谱法 Gel-permeation chromatography HA 酸性溶质,能电离出A- Hex 己烷 Hexane hr 二相邻谱带之间的谷高 HVA 高香草酸 Homovanillic acid h’ 峰高 h1,h2 相邻谱峰1和谱峰2的峰高 IEC 离子交换色谱法 Ion-exchange chromatography IP 离子对 Ion-pair IPC 离子对色谱法 Ion-pair chromatography J 色谱峰强度参数 K’ 所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’) k 梯度洗脱过程中,某溶质的k’的平均值或有效值 kw 以水做流动相k’的外推值 k1,k2 相邻谱峰1和谱峰2的容量因子 L 色谱柱长度(cm) Lc 检测器流动池光路的长度(cm) M 溶质的分子量 MC 二氯甲烷 Methylene chloride MDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件 MeOH 甲醇 Methanol MTBE 甲基叔丁醚 Methyl-t-butyl ether MW 溶质的分子量 N 色谱柱塔板数 NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质) N0 检测器的基线噪音 ODS 十八烷基硅烷 Octadecylsilyl P 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数 PA 普鲁卡因胺 Procainamide(碱性物质) PAH 聚芳香烃 Polyaromatic Hydrocarbon PESOS 优化流动相的计算机软件(美国Perkin-Elmer产品) pKa 溶质酸性常数的负对数;当pH=pKa时,溶质中有一半是电离的 Rk 保留值范围,Rk=(最末谱峰k’)/(最初谱峰k’) RRM 相对分离度图(通常N=10000) Rs 相邻二谱峰的分离度 S 当流动相中的%B改变时,测量溶质保留值的变化速率的参数 SAL 水杨酸 Salicylic Acid SEC 尺寸排阻色谱法 Size-exclusion chromatography S/N 信噪比 Signal to noise ratio t 分离时间(min)(样品进样时t=0) tp 梯度系统的滞后时间(min) TBA 四丁基铵离子 Tetrabutylammonium ion TEA 三乙胺 Triethylamine THF 四氢呋喃 Tetrahydrofuran tk 在用于校正等度洗脱溶剂强度的流动相离开梯度混合器时,梯度洗脱的时间 TLC 薄层色谱法 Thin-layer chromatography TMA 四甲基铵 Tetramethylammonium(盐) TMS 三甲基硅烷 Trimethylsilyl t0 色谱柱的死时间(min) tR 溶质的保留时间(min) tG 梯度时间(min),即梯度开始至结束的时间 t1,t2 相邻谱峰1和谱峰2的保留时间(min) ti 色谱图中第一峰的保留时间(min) tf 色谱图中最末峰的保留时间(min) △tg tf-ti tx (tf-ti)/2 UV 紫外光 Vm 色谱柱的死体积(mL),Vm=t0F VMA 香草扁桃酸 Vanillymandelic acid wm 化合物的进样量 w1,w2 相邻谱峰1和谱峰2于半峰高处(W1/2)的宽度(min) W1,W2 相邻谱峰1和谱峰2的基线宽度(min) W1/2 半峰高处的谱带宽度 xd,xe,xn 溶剂选择参数,分别用于测定溶剂的酸度、碱度和偶极性的程度 α 分离因子,α=k2/k1 △Φ 梯度洗脱期间流动相成分的变化 εo 溶剂强度参数 ε 化合物的克分子吸收系数 η 流动相的粘度(Pas) Φ 流动相中强溶剂的体积份数 %B 二元流动相中强溶剂的体积百分比(%v)
作者:兰文; 杨汉初; 黄莉;(湖南省药品检验所;)摘要:目的建立反相高效液相色谱法测定复方明矾布比卡因注射液的有关物质及盐酸布比卡因的含量。方法采用Diamonsil C18柱,以乙腈-磷酸盐缓冲液(取磷酸二氢钾1.94g,磷酸氢二钾2.48g,加水溶解并稀释至1 000mL,调节pH 6.8)(65∶35)为流动相;流速为1.0mL.min-1;有关物质检测波长为215nm,含量测定检测波长为263nm;柱温:30℃。结果在该色谱条件下,杂质峰与主峰均能有效分离,盐酸布比卡因在44.31~177.26μg.mL-1与峰面积线性关系良好(r2=0.999 7);平均回收率为99.7%(n=9),RSD=0.4%。结论本法简便、快速、准确,专属性好,可用于复方明矾布比卡因注射液的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061043_381724_1606903_3.jpg
液相色谱常用符号与术语表ACN 乙腈 AcetonitrileAUFS 满量程的吸光度单位 Absorbance units, full scaleAs 峰不对称因子B 二元流动相中的强溶剂;例如:反相HPLC的甲醇/水混合液中的甲醇BSA 牛血清白蛋白(一种蛋白质) Bovine serum albuminCAF 咖啡因(中性溶质) CaffeineCRF 色谱响应因子 Chromatographic response function;色谱图总分离度的定量指标dc 色谱柱内径(cm)DMOA 二甲基辛胺 DimethyloctylamineDNB 2,4-二硝基甲酰(基) 2,4-Dinitrobenzoyldp 色谱柱填料的粒度(cm)DRYLAB 液相资源公司(LC Resources INC.)的计算机模拟软件。DRYLAB I用于等度预测,DRYLAB G用于梯度预测F 流动相的流速(ml/min)FC-113 1,1,2-三氟-1,2,2-三氯乙烷GPC 凝胶渗透色谱法 Gel-permeation chromatographyHA 酸性溶质,能电离出A-Hex 己烷 Hexanehr 二相邻谱带之间的谷高HVA 高香草酸 Homovanillic acidh’ 峰高h1,h2 相邻谱峰1和谱峰2的峰高IEC 离子交换色谱法 Ion-exchange chromatographyIP 离子对 Ion-pairIPC 离子对色谱法 Ion-pair chromatographyJ 色谱峰强度参数K’ 所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’)k 梯度洗脱过程中,某溶质的k’的平均值或有效值kw 以水做流动相k’的外推值k1,k2 相邻谱峰1和谱峰2的容量因子L 色谱柱长度(cm)Lc 检测器流动池光路的长度(cm)M 溶质的分子量MC 二氯甲烷 Methylene chlorideMDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件MeOH 甲醇 MethanolMTBE 甲基叔丁醚 Methyl-t-butyl etherMW 溶质的分子量N 色谱柱塔板数NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质)N0 检测器的基线噪音ODS 十八烷基硅烷 OctadecylsilylP 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数PA 普鲁卡因胺 Procainamide(碱性物质)PAH 聚芳香烃 Polyaromatic HydrocarbonPESOS 优化流动相的计算机软件(美国Perkin-Elmer产品)pKa 溶质酸性常数的负对数;当pH=pKa时,溶质中有一半是电离的Rk 保留值范围,Rk=(最末谱峰k’)/(最初谱峰k’)RRM 相对分离度图(通常N=10000)Rs 相邻二谱峰的分离度S 当流动相中的%B改变时,测量溶质保留值的变化速率的参数SAL 水杨酸 Salicylic AcidSEC 尺寸排阻色谱法 Size-exclusion chromatographyS/N 信噪比 Signal to noise ratiot 分离时间(min)(样品进样时t=0)tp 梯度系统的滞后时间(min)TBA 四丁基铵离子 Tetrabutylammonium ionTEA 三乙胺 TriethylamineTHF 四氢呋喃 Tetrahydrofurantk 在用于校正等度洗脱溶剂强度的流动相离开梯度混合器时,梯度洗脱的时间TLC 薄层色谱法 Thin-layer chromatographyTMA 四甲基铵 Tetramethylammonium(盐)TMS 三甲基硅烷 Trimethylsilylt0 色谱柱的死时间(min)tR 溶质的保留时间(min)tG 梯度时间(min),即梯度开始至结束的时间t1,t2 相邻谱峰1和谱峰2的保留时间(min)ti 色谱图中第一峰的保留时间(min)tf 色谱图中最末峰的保留时间(min)△tg tf-titx (tf-ti)/2UV 紫外光Vm 色谱柱的死体积(mL),Vm=t0FVMA 香草扁桃酸 Vanillymandelic acidwm 化合物的进样量w1,w2 相邻谱峰1和谱峰2于半峰高处(W1/2)的宽度(min)W1,W2 相邻谱峰1和谱峰2的基线宽度(min)W1/2 半峰高处的谱带宽度xd,xe,xn 溶剂选择参数,分别用于测定溶剂的酸度、碱度和偶极性的程度α 分离因子,α=k2/k1△Φ 梯度洗脱期间流动相成分的变化εo 溶剂强度参数ε 化合物的克分子吸收系数η 流动相的粘度(Pas)Φ 流动相中强溶剂的体积份数%B 二元流动相中强溶剂的体积百分比(%v)
ACN 乙腈 Acetonitrile AUFS 满量程的吸光度单位 Absorbance units, full scale As 峰不对称因子 B 二元流动相中的强溶剂;例如:反相HPLC的甲醇/水混合液中的甲醇 BSA 牛血清白蛋白(一种蛋白质) Bovine serum albumin CAF 咖啡因(中性溶质) Caffeine CRF 色谱响应因子 Chromatographic response function;色谱图总分离度的定量指标 dc 色谱柱内径(cm) DMOA 二甲基辛胺 Dimethyloctylamine DNB 2,4-二硝基甲酰(基) 2,4-Dinitrobenzoyl dp 色谱柱填料的粒度(cm) DRYLAB 液相资源公司(LC Resources INC.)的计算机模拟软件。DRYLAB I用于等度预测,DRYLAB G用于梯度预测 F 流动相的流速(ml/min) FC-113 1,1,2-三氟-1,2,2-三氯乙烷 GPC 凝胶渗透色谱法 Gel-permeation chromatography HA 酸性溶质,能电离出A- Hex 己烷 Hexane hr 二相邻谱带之间的谷高 HVA 高香草酸 Homovanillic acid h’ 峰高 h1,h2 相邻谱峰1和谱峰2的峰高 IEC 离子交换色谱法 Ion-exchange chromatography IP 离子对 Ion-pair IPC 离子对色谱法 Ion-pair chromatography J 色谱峰强度参数 K’ 所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’) k 梯度洗脱过程中,某溶质的k’的平均值或有效值 kw 以水做流动相k’的外推值 k1,k2 相邻谱峰1和谱峰2的容量因子 L 色谱柱长度(cm) Lc 检测器流动池光路的长度(cm) M 溶质的分子量 MC 二氯甲烷 Methylene chloride MDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件 MeOH 甲醇 Methanol MTBE 甲基叔丁醚 Methyl-t-butyl ether MW 溶质的分子量 N 色谱柱塔板数 NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质) N0 检测器的基线噪音 ODS 十八烷基硅烷 Octadecylsilyl P 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数 PA 普鲁卡因胺 Procainamide(碱性物质) PAH 聚芳香烃 Polyaromatic Hydrocarbon PESOS 优化流动相的计算机软件(美国Perkin-Elmer产品) pKa 溶质酸性常数的负对数;当pH=pKa时,溶质中有一半是电离的 Rk 保留值范围,Rk=(最末谱峰k’)/(最初谱峰k’) RRM 相对分离度图(通常N=10000) Rs 相邻二谱峰的分离度 S 当流动相中的%B改变时,测量溶质保留值的变化速率的参数 SAL 水杨酸 Salicylic Acid SEC 尺寸排阻色谱法 Size-exclusion chromatography S/N 信噪比 Signal to noise ratio t 分离时间(min)(样品进样时t=0) tp 梯度系统的滞后时间(min) TBA 四丁基铵离子 Tetrabutylammonium ion TEA 三乙胺 Triethylamine THF 四氢呋喃 Tetrahydrofuran tk 在用于校正等度洗脱溶剂强度的流动相离开梯度混合器时,梯度洗脱的时间 TLC 薄层色谱法 Thin-layer chromatography TMA 四甲基铵 Tetramethylammonium(盐) TMS 三甲基硅烷 Trimethylsilyl t0 色谱柱的死时间(min) tR 溶质的保留时间(min) tG 梯度时间(min),即梯度开始至结束的时间 t1,t2 相邻谱峰1和谱峰2的保留时间(min) ti 色谱图中第一峰的保留时间(min) tf 色谱图中最末峰的保留时间(min) △tg tf-ti tx (tf-ti)/2 UV 紫外光 Vm 色谱柱的死体积(mL),Vm=t0F VMA 香草扁桃酸 Vanillymandelic acid wm 化合物的进样量 w1,w2 相邻谱峰1和谱峰2于半峰高处(W1/2)的宽度(min) W1,W2 相邻谱峰1和谱峰2的基线宽度(min) W1/2 半峰高处的谱带宽度 xd,xe,xn 溶剂选择参数,分别用于测定溶剂的酸度、碱度和偶极性的程度 α 分离因子,α=k2/k1 △Φ 梯度洗脱期间流动相成分的变化 εo 溶剂强度参数 ε 化合物的克分子吸收系数 η 流动相的粘度(Pas) Φ 流动相中强溶剂的体积份数 %B 二元流动相中强溶剂的体积百分比(%v)
[size=5][b]科学、高效、安全地使用兽药,不但能及时预防和治疗动物疾病,提高农户养殖效益,而且对控制和减少药物残留,提高动物产品品质等具有重要意义。使用兽药应注意如下五个问题。 充分考虑药物的特性 内服能吸收的药物,可以用于全身感染;内服不能吸收的药物,只能用于胃肠道感染。 选择合适的用药途径 苦味健胃药如龙胆酊、马钱子酊等,只有通过口服的途径,才能刺激味蕾,加强唾液和胃液的分泌,如果使用胃管投药,就起不到健胃的作用。 注意药物的有效浓度 肌肉注射卡那霉素,有效浓度维持时间为12小时,因此,连续肌肉注射卡那霉素,间隔时间应在10小时以内。青霉素粉针剂一般应每隔4小时~6小时重复用药1次,油剂普鲁卡因青霉素则可以间隔24小时用药1次。 尽量选用效能多样或有特效的药物 幼畜禽发生黄痢、白痢时,尽早选用黄连素;安普霉素治疗家禽的大肠杆菌、沙门氏杆菌感染,疗效非常显著。 注意药物之间的配伍禁忌酸性药物与碱性药物不能混合使用;口服活菌制剂时应禁用抗菌药物和吸附剂;磺胺类药物与维生素C合用,会产生沉淀;磺胺嘧啶钠注射液与大多数抗生素配合都会产生浑浊、沉淀或变色现象,应单独使用。[/b][/size]
辐射灭菌是诸多灭菌方法中的一种,但那些药物适合用辐射法灭菌?那些药物不适合用辐射法灭菌呢?现简要总结,供同一个战壕的战友参考。一、可用辐射法灭菌的药物包括:1、抗菌素类:青霉素钾、普卡因青霉素水混悬液、普鲁卡因青霉素油剂、苯基青霉素钠、苯甲氧基青霉素、氨苄青霉素、链霉素、氯霉素、红霉素、卡那霉素、金霉素、土霉素、万古霉素、硫酸新霉素、制菌霉素、利福平、两性霉素B等 2、激素类:丙酸睾丸素粉液及其油溶液、雌二醇、己烯雌酚、醋酸孕烯炔醇酮、孕甾醇、醋酸皮质素、可的松、强的松龙 3、巴比妥类:苯巴比、苯巴比妥、戊巴比妥、戊巴比妥4、菌苗抗毒素类 :斑疹伤寒疫苗、白喉抗毒素 5其他类:生理盐水、注射用水、盐酸普鲁卡因及其溶液、10%葡萄糖酸钙 二、不适宜用辐射法灭菌的药物包括:1、生物碱类:硫酸阿托品注射液、盐酸吗啡注射液、盐酸麻黄碱注射液等、苹果酸麦角新碱注射液、0.5%盐酸利多卡因等。2、生物制品类:脑垂体后叶激、胰岛素注射液、果青蛋白锌胰岛素注射液、肝素及其注射液、果青制破伤风抗毒素、三磷酸腺苷注射液、冻干人血浆 等。3、强心苷类:毛花强心苷注射液、洋地黄毒甙注射液等 4、维生素类:维生素B6液、维生素B12注射液等 5、酶类:玻璃酸酶、粘脘酸 等
高效液相色谱有四方面重要的作用。①分离分析药物的组分含量:由于一种药剂常含有多组分,色谱方法是既能分离又能定量的方法。如脂溶性维生素片,含有铬维生素,有C18柱及含1%碳酸铵的甲醇为流动相。一次即可将各维生素B6、D1、A、E同时分离并定出含量。又如止痛药或退烧药也可用此法分离并测得各组分的含量。 ②药物生产中进行中间控制:生产抗菌素时,首先要了解发酵液中的主体与付产物的存在情况。例如:青霉素、四环素、头孢子菌素、红霉素、柔红霉素、庆大霉素等的发酵产物,均可用C18柱和甲醇及缓冲液配制的流动相或用离子对法,分析主体及其收产物的含量。对普鲁卡因酰胺、茶碱等可进行主体含量的测定,对硝酸甘油可测定主体含量及其降解产物,以便保证药物的热量。 ③分析药物在体内的残量:这是近来研究新药效能很主要的内容之一,如冬凌草甲素是植物中的一组分,有抗癌效果,但是毒性大。测定其在血清中含量很重要,现在可用C18柱及甲醇水为流动相,测定处理后的血清样品,即可得到血中的含量。如磺胺类药品在血清及尿中含量也不允许太大,可用C18柱及酸性乙醇为流动相,测定含量后,则可估计最合适的用药剂量。又如四环素、巴比妥酸、止痛药、退烧药、护糖尿病的药物等,在血或尿中的含量都可用C18柱用甲醇、水为流动相来测定含量。因此,反相色谱法应用范围极广。 ④测定药物在各种器官中的代谢产物,特别是研究药物的代谢产物在血清中存在的情况。如测血清中的酮酸及戊二酸。测尿中代谢产物更为方便,不需要繁锁的预处理。因为研究尿中的组分能鉴别病人的代谢功能,对病理研究或监床诊断均具有重要的参考价值。
[color=black]复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[/color][color=black] [/color][img=,156,99]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211036489419_4502_2297_3.jpg!w156x99.jpg[/img][align=center][/align][align=left][b][color=black]盐酸阿替卡因(Articaine hydrochloride M.W.:320.84)[/color][/b][/align][align=center][b][color=black] [/color][/b][/align][color=black]在现有国家药品标准(YBH17082004-2015Z)分析方法中,流动相添加了离子对试剂-庚烷磺酸钠,并在pH为2.0的强酸条件下进行相应分析,不利于色谱柱的使用寿命。大曹三耀实验室参考USP方法,以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,实现了复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析(复方盐酸阿替卡因注射液由客户提供)。[/color][color=black]CAPCELLPAK C18 MGII[/color][color=black]液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。[/color][align=left][b][color=#0070c0]实验方法[/color][/b][/align][align=left][img=,500,358]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211037456969_5082_2297_3.jpg!w730x523.jpg[/img][/align][align=left]图1[color=black]盐酸阿替卡因[/color]对照品及供试品溶液[/align][align=left][img=,500,248]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211038541919_2603_2297_3.jpg!w572x284.jpg[/img][/align][align=center][/align][align=center][/align][color=black]为进行有关物质分析,该实验将注射液样品以流动相稀释100倍,作为有关物质供试品溶液,再将该有关物质供试品溶液以流动相进一步稀释100倍,作为自身对照溶液。以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,通过调整流动相比例及柱温,最终在18%乙腈、柱温30℃条件下实现了盐酸阿替卡因供试品溶液及对照品的良好分析。[/color]如图2、3,使用CAPCELL PAK C18 MGII色谱柱进行分析,盐酸阿替卡因和有关物质均能得到良好分析结果,主峰与峰前杂质得到了良好分离,分离度为1.90(见表1)[img=,400,311]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045536855_9516_2297_3.jpg!w574x447.jpg[/img][img=,400,295]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045540875_8483_2297_3.jpg!w698x516.jpg[/img][align=left] 图2 [color=black]盐酸阿替卡因[/color]有关物质供试品溶液及空白 图3 自身对照溶液[/align][align=center][/align][align=left]表1 有关物质结果详表[/align][align=left][img=,600,323]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211042513275_6690_2297_3.jpg!w786x424.jpg[/img][/align][align=center][/align]综上实验结果,使用CAPCELL PAK C18 MGII S5 4.6mm i.d.×250 mm色谱柱,以冰醋酸水溶液-乙腈为流动相体系,在30°C柱温条件下,能够实现复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析。[color=black] [/color]
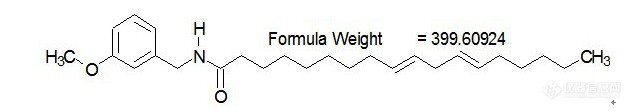
公司玛咖研究团队专心致力于我国玛咖产业发展,团队核心成员多年来一直致力于玛咖在我国的产业化推进,尤其是通过长期的研究玛咖药效物质基础和作用机制,发掘出多种具有医药健康产品开发潜力的玛咖生物活性成分,如:玛咖酰胺,玛咖烯,芥子油苷等,并开展玛咖生物活性成分含量及基础营养成分分析——标准建立、玛咖品质评价、深加工产品开发、玛咖活性物质基础研究——玛咖提取物及深加工产品开发、医药产品开发。 现应部分玛咖科研机构及深加工企业需求,提供部分玛咖酰胺标准品,信息如下:[color=#ff00ff] 中文名:[/color][color=#ff00ff]间-甲氧基苄基-[/color][color=#ff00ff]亚麻酰胺[/color];N-(3-甲氧基苄基)-(9Z, 12Z, 15Z)-十八碳三烯酰胺 英文名:N-(3-methoxybenzyl)-(9Z,12Z,15Z)-octadecatrienamide 分子式:C26H39NO2 [color=#ff00ff]CAS:[url=https://detail.1688.com/offer/565181222786.html?spm=a2615.7691456.0.0.46631c53wILZes#][color=#ff00ff]883715-23-9[/color][/url][/color] 分子量:397 贮存条件: -20℃冷藏、密封、避光 推荐溶剂:甲醇、乙腈 纯 度: ≥ 95% 用 途: 用于含量测定/鉴定/药理实验等。 结构式:[img=,633,111]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640131086_801_3030551_3.jpg!w633x111.jpg[/img] 规 格:20mg 含 量:98% 储存温度:0℃~8℃[img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640324296_653_3030551_3.jpg!w690x490.jpg[/img] 公司玛咖研究团队专心致力于我国玛咖产业发展,团队核心成员多年来一直致力于玛咖在我国的产业化推进,尤其是通过长期的研究玛咖药效物质基础和作用机制,发掘出多种具有医药健康产品开发潜力的玛咖生物活性成分,如:玛咖酰胺,玛咖烯,芥子油苷等,并开展玛咖生物活性成分含量及基础营养成分分析——标准建立、玛咖品质评价、深加工产品开发、玛咖活性物质基础研究——玛咖提取物及深加工产品开发、医药产品开发。[img=,690,370]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640463341_1492_3030551_3.jpg!w690x370.jpg[/img] 玛咖研发技术服务: 玛咖研究中心引领我国玛咖产业发展——玛咖核心技术研究——玛咖良种培育——玛咖标准化种植——采收及深加工——玛咖新型健康产品开发和推广。 核心技术:玛咖良种培育——玛咖标准化种植——玛咖中生物活性成分含量检测分析——玛咖中活性成分浓缩提取制备技术——玛咖生理活性物质作用机理研究——玛咖深加工过程中生物活性成分稳定性研究等。[img=,690,387]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641093953_8734_3030551_3.jpg!w690x387.jpg[/img] 开展多项技术服务: 玛咖研究中心实现了玛咖中生物活性成分物质,如:玛咖酰胺、玛咖烯、玛咖咪唑生物碱、玛咖苄基芥子油苷、异硫氰酸苄酯、腺苷类物质等玛咖特征活性物质的结构解析、含量测定、制备技术。 玛咖样品检测分析: 玛咖生物活性成分:1 、玛咖酰胺;2、 玛咖烯;3、 总芥子油苷;4、 挥发油(主要检测异硫氰酸苄酯类物质等);5 、玛咖咪唑生物碱;6 、甾醇 ;7、 皂苷;8、 腺苷等 玛咖基础营养成分:1 、蛋白质;2、 氨基酸(氨基酸总量及水解17种氨基酸比例);3 、膳食纤维等 [color=#3333ff] [b]玛咖研发中心(武汉)面向全国提供玛咖检测服务[/b][/color] 玛咖是一种原产南美安第斯山脉海拔3800米处的高原珍稀药食两用植物。在秘鲁,当地人把玛咖奉为“安第斯皇后”,古印加帝国的“皇室贡品”,从20世纪60年开始,多国科研人员对玛咖作用进行临床试验,证明:玛咖含有多种丰富,均衡合理的营养成分及多种生理活性物质,通过调节内分泌系统平衡,达到提高生育力、抗疲劳的目的,被誉为天然的“荷尔蒙发动机”,因为玛咖能够温和的调理身体机能,这些独有的功能是其它药剂所不能替代的。 2005年前后玛咖在我国形成一定规模。由于玛咖品种的差异、种植技术的差异、玛咖产地的差异、加工方式的差异等,我国玛咖原料和产品的营养成分和活性成分也就有一定差异,这就影响了玛咖的食用效果和产品品质稳定性,因此无论是玛咖种植基地、原料生产企业还是玛咖产品深加工企业,都有必要对玛咖品质进行全面的检测分析。[img=,690,339]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641268751_4701_3030551_3.jpg!w690x339.jpg[/img] 玛咖品质优劣主要包括生物活性成分和营养成分两部分,玛咖生物活性成分则应该关注:玛咖酰胺、玛咖烯、玛咖芥子油苷等成分,营养成分中应该关注:玛咖粗蛋白质、17种氨基酸含量及比例、膳食纤维和矿物质等,这些成分的品质分析及含量检测对于一些普通企业来说有较大困难,尤其是活性成分检测需要用到[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url](HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url])、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url](HPLC-GS)等高新技术手段。为方便玛咖种植基地、原料供应商及玛咖产品深加工企业全面了解玛咖品质,本公司现面向全国提供玛咖品质分析及检测服务。 [img=,690,349]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641415015_6891_3030551_3.jpg!w690x349.jpg[/img] 武汉华士特玛咖研发中心科研人员长期致力于玛咖研究与产业化开发工作,已经为秘鲁( Perú )利马、玻利维亚、厄瓜多尔;云南丽江、云南香格里拉、云南昭通、云南宣威;西藏林芝、西藏曲水、西藏昌都;新疆塔什库尔干、喀什、四川阿坝;宁夏、青海;吉林抚松等多地玛咖种植基地、原料供应商及产品深加工企业提供了玛咖检测服务,年均服务玛咖种植基地及玛咖产品开发企业超过300家,接待样品检测量超过1000份,同时建立了玛咖研究中心全球玛咖样本及产品展示中心,同时依托玛咖分析测试中心首次建立了国内、外玛咖品质大数据库,追溯国内外各玛咖产区的品质变迁,为玛咖研究和产业化开发提供技术支撑。同时,玛咖研究中心是目前国内玛咖成分检测服务最全面的单位。[img=,690,304]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121642040581_2553_3030551_3.jpg!w690x304.jpg[/img] 检测服务项目包括:玛咖品种鉴定和玛咖产品真伪辨别、玛咖DNA指纹图谱鉴定、玛咖营养成分分析(总蛋白、氨基酸、膳食纤维、总糖、矿物质等)、玛咖活性成分分析(玛咖酰胺、玛咖烯、玛咖芥子油苷、玛咖挥发油、玛咖甾醇、玛咖咪唑生物碱、腺苷等)、各类玛咖产品活性成分分析(玛咖根、玛咖干粉、玛咖片、玛咖胶囊、玛咖片剂、玛咖口服液、玛咖提取物、玛咖饮料、玛咖酒)、玛咖中常见违禁物添加筛查(可对玛咖产品里添加的西地那非、他达那非及其类似物进行分析)、其它检测项目。 检测费用:通过电话咨询,提供专业性的检测技术服务,同时可与种植基地和产品开发企业签订长期检测技术服务协议,为种植基地玛咖品质和玛咖产品开发企业提供优质的品控服务; 检测周期:7-30日,具体需根据检测项目和数量确定; 检测咨询网址:[url]https://huaster.1688.com/[/url] QQ:422450190 电话:15926423062
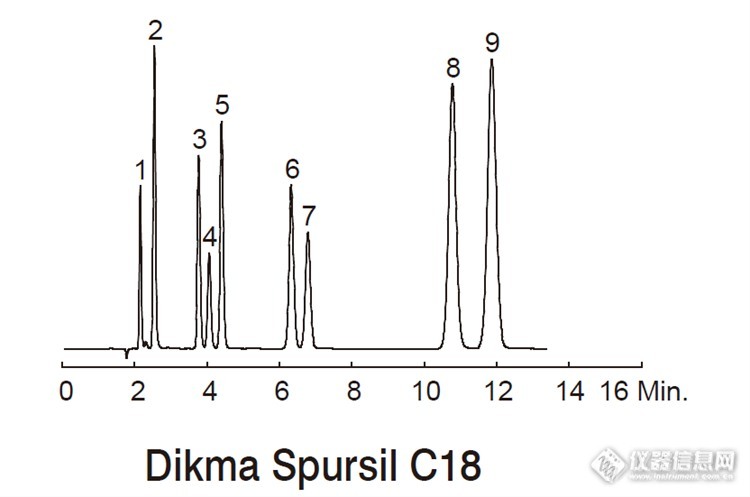
咖啡的主要成分是咖啡因,可以作用于神经细胞中一种叫做腺嘌呤核苷的化学物质,是一种中枢神经兴奋剂,能够暂时的驱走睡意并恢复精力。 不过,咖啡对有些人是有好处的,但是对某些人却产生负面影响,这主要是与咖啡因的在不同人的代谢能力有关的。 咖啡因在肝脏中被分解产生三个初级代谢产物副黄嘌呤,可可碱,茶碱。咖啡因在摄取后45分钟内被胃和小肠完全吸收。吸收后它会分布于身体的所有器官之中,转化过程符合化学动力学一级反应,这些化合物进一步代谢,最终通过尿液排泄。 如果某些人的这个酶的代谢比较快,摄入的咖啡因很快就会被清除出体外,因此咖啡因起作用的效果就很有限,不能令人产生特别明显的兴奋感。而对于另一些人,他们这个酶代谢速度慢,咖啡因在体内的清除速度很慢,起作用时间也就较长,这样的人往往一杯咖啡就会令他们夜不能寐,有的还会影响食欲,呕吐和痉挛,也可能出现胃炎或心脏病等不良反应。 所以这也解释了一般人普遍担心的咖啡会影响睡眠问题,其实和你对咖啡因的代谢力有很大的关系。由于每个人对咖啡因代谢的能力不同,平均来说,咖啡因在体内的运作,大约能维持3~4个小时,所以即使在晚餐后饮用,也不至于造成太大的困扰。但有些人的代谢力较差,可能会持续作用8~12个小时;或者体质对咖啡因比较敏感,就得特别注意喝咖啡的时间,避免影响作息,因为不论是多喝开水或是增加运动,都无法有效的促使咖啡因快速代谢。*********************************** 以上对咖啡因的代谢方面做了简单的科普,可如何对咖啡因代谢物进行检测呢?由于咖啡因代谢物从化学结构上来看,这一类化合物具有相似的母体结构,不同之处在于甲基的位置,属于位置异构体。(见下图)http://ng1.17img.cn/bbsfiles/images/2016/01/201601071109_581149_2452211_3.png 对于分离这些代谢物来说,对色谱柱的分离能力要求比较高,下面我们看看使用迪马Spursil色谱柱分离9种咖啡因代谢物的分离效果情况色谱柱:Spursil C18规格:150 x 4.6 mm, 5 μm流动相:甲醇/ 水+1% 乙酸=10/90流速:1.0 mL/min柱温:室温检测器:UV 254 nm样品:1. 尿酸2. 黄嘌呤3. 7- 甲基黄嘌呤4. 1- 甲基尿酸5. 3- 甲基黄嘌呤6. 1,3- 二甲基尿酸7. 可可碱8. 1,7- 二甲基黄嘌呤9. 茶碱http://ng1.17img.cn/bbsfiles/images/2016/01/201601071110_581151_2452211_3.png总结:Spursil 色谱柱能够在13分钟之内将它们全部分开且达到基线分离~棒棒哒http://simg.instrument.com.cn/bbs/images/default/em09505.gif
各位高手可有盐酸罗哌卡因红外谱图啊?还有顺便问下,药典上说药品1-2mg氯化钾200mg,其中[color=#d40a00]1-2mg[/color]和[color=#d40a00]200mg[/color]只是个用质量来描述固体样品量的方式吧,概数,无需用天平来称量、量化吧?正如液体样品可用体积的量来描述,L,ml或滴。是这样理解的吧?
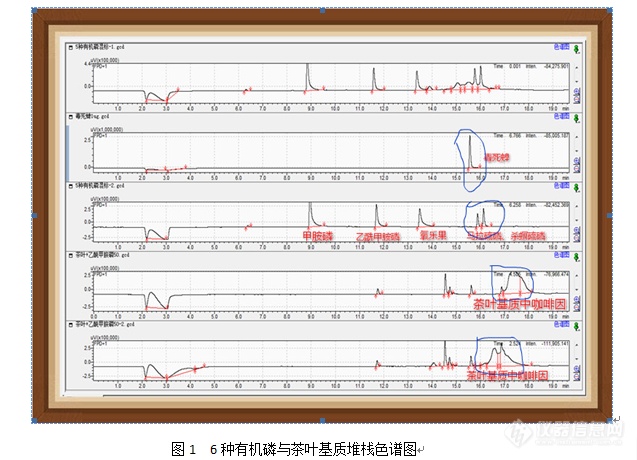
茶叶中咖啡因对有机磷类农药残留测定的影响摘要:茶叶中经常要检测有机磷类农药(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷),由于茶叶样品基质富含咖啡因的特殊性,本文主要阐述了用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定茶叶中这些有机磷时,咖啡因对这些农药测定的影响,及如何准确地测定这些有机磷农药残留。关键词:茶叶;咖啡因;有机磷农药;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法;准确定量前言:茶叶是深受人们喜爱的一种饮料这一,最近来,滥用农药的现象也越来越严重,GB 2763-2016[sup][/sup]也对茶叶中各种农药残留的限量作了具体的规定。众所周知,茶叶富含芳香族化合物、多酚和咖啡因[sup][/sup]。咖啡因等化合物会在前处理的萃取过程中与农药残留一起被萃取出来,如果没有有效地去除,将会对目标农药(毒死蜱、马拉硫磷、杀螟硫磷)的准确定量造成影响,同时有机磷类农药(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷)用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析时通常会一起进行净化前处理,一起上机用FPD检测器上机分析。这又给部分农药残留(甲胺磷、乙酰甲胺磷、氧乐果)的准确定量造成了失误。实验仪器:岛津[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] -2010plus(配FPD); 色谱柱:RTX-170130m*0.25mm,0.25um[align=left]仪器条件:进样口温度:220 ℃ 检测器温度:230 ℃[/align]程序升温:[color=black]60℃[/color][color=black]([/color][color=black]1 min[/color][color=black])[/color][sup][color=black]20[b]℃[/b]/min [/color][/sup][color=black]150℃ [sup]15[b]℃[/b]/min[/sup] 230℃ [sup]25[b]℃[/b]/min[/sup] 280℃[/color][color=black]([/color][color=black]5 min[/color][color=black])[/color][color=black][/color]标准品:甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷、[color=red]咖啡因[/color]样品处理(方法一):依SN/T1950-2007[sup][/sup]对茶叶进行处理,同时进行加标实验。提取:取样1.0g(±0.01g)于50mL塑料离心管中,加入1mL饱和氯化钠水溶液浸泡十分钟左右,加入15mL乙酸乙酯先均质,再加入一勺无水硫酸钠和2勺无水硫酸镁均质30s,用15mL乙酸乙酯洗均质头合并提取液,盖上盖子振摇一会,超声5min。把提取液和残渣一起直接过加有2勺无水硫酸镁的漏斗入鸡心瓶中,2×5mL乙酸乙酯洗离心管,振摇,合并提取液于鸡心瓶中。再用20mL乙酸乙酯冲洗漏斗上的残渣合并洗液,35℃旋转蒸发至剩2mL左右,待净化。净化:10mL丙酮+正己烷(1+1, V+V)先活化TPT(10mL,2g)柱子(填料上加1cm左右高的无水硫酸钠)下接15mL玻璃离心管,将上述大约剩2mL左右的乙酸乙酯先吸出直接过活化后的TPT柱,再用丙酮+正己烷(1+1, V+V)2.5 mL×2次洗涤鸡心瓶(必要时超声波),洗液继续过TPT柱子,加丙酮+正己烷(1+1, V+V)继续淋洗柱子共收集12mL淋洗液,35℃左右氮气吹干,丙酮+正己烷(1+1, V+V)定容1mL上机测试。 混标(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷)与茶叶基质堆栈色谱图,如图1所示:[img=,637,460]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161355391717_7304_2166779_3.png!w637x460.jpg[/img]从图1中可以看出:茶叶基质(空白茶叶)在毒死蜱、马拉硫磷、杀螟硫磷出峰位置附近出现很大的坡(后经确认是茶叶基质中的咖啡因:[color=red]前处理浓缩定容时会产生白色絮状物,此白色絮状物就是茶叶基质中的咖啡因析出产生的[/color]),给茶叶中毒死蜱、马拉硫磷、杀螟硫磷准确定量带来了严重影响。[color=red]且[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]进了茶叶基质中的咖啡因后,要连续进十几针的丙酮空白针,此坡才能消失,才不会对后续的出峰位置的物质定量产生干扰。[/color][color=red][/color]样品处理(方法二):(目标:要去除茶叶基质中的咖咖啡因。)[color=red][/color][align=left]提取:取样2.0g(±0.01g)于50mL塑料离心管中,加入2mL去离子水、20mL丙酮+正己烷(1+1,V+V)溶液和一勺无水硫酸钠,旋紧离心管盖,涡旋1min后超声30min,超声期间每5min振摇一次,4000 r/min离心5min,待净化。[/align]净化: 移取5.0mL上清液至15mL离心管中,35℃下氮气吹干,加入2.5mL正己烷涡旋使样品溶解,[color=red]再加入[/color][color=red]2.5mL[/color][color=red]饱和氯化钠水溶液继续涡旋[/color][color=red]30s[/color]说明:[color=red]用饱和氯化钠水溶液和正己烷分配可去除水溶性杂质及咖啡因)[/color]后2000 r/min离心1min,取出正己烷层,剩余溶液中加入2.5mL正己烷再提出一次。合并正己烷层过经5mL丙酮+正己烷(1+1,V+V)活化上填1cm高无水硫酸钠的Carb/PSA柱(0.5g/ 6mL),用8mL丙酮+正己烷(1+1,V+V)继续洗脱,共收集洗脱液13mL于15mL刻度玻璃离心管中,35℃水浴氮气吹干,用正己烷定容1mL上[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-FPD检测。同时进行加标处理(加标量:加入200ng/mL的有机磷混标1mL,与样品同时同样处理,相当于最终上机浓度为40ng/mL)。从图1中可以看出:茶叶基质(空白茶叶)在毒死蜱、马拉硫磷、杀螟硫磷出峰位置附近出现很大的坡(后经确认是茶叶基质中的咖啡因:[color=red]前处理浓缩定容时会产生白色絮状物,此白色絮状物就是茶叶基质中的咖啡因析出产生的[/color]),给茶叶中毒死蜱、马拉硫磷、杀螟硫磷准确定量带来了严重影响。[color=red]且[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]进了茶叶基质中的咖啡因后,要连续进十几针的丙酮空白针,此坡才能消失,才不会对后续的该出峰位置附近的物质定量产生干扰。[/color][color=red][/color][img=,690,415]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161358058714_1404_2166779_3.png!w690x415.jpg[/img][img=,663,266]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161358110432_4139_2166779_3.png!w663x266.jpg[/img] 表1 用方法二处理各有机磷的回收率从图2及表1可以看出:使用方法二处理([color=red]用饱和氯化钠水溶液和正己烷分配可去除水溶性杂质及咖啡因:茶叶样品基质中的咖啡因大谷峰[/color][color=red]已消失[/color])毒死蜱、马拉硫磷、杀螟硫磷可以准确地给予定量了,然而甲胺磷、乙酰甲胺、氧乐果却也同时会与咖啡因从正己烷层进入饱和的氯化钠层而被洗脱除去。因此用方法二来处理茶叶基质的方法可以很好地去除咖啡因基质,适用于检测毒死蜱、马拉硫磷、杀螟硫磷,不适用于同时要检测:甲胺磷、乙酰甲胺、氧乐果这三种有机磷的净化方法。(必要时)样品处理(方法三):适用于茶叶中甲胺磷,乙酰甲胺磷的净化方法。取干样0.5g于50mL塑料离心管中加2mL水,放置至少30分钟。加入20mL乙酸乙酯和10g无水硫酸钠,均质0.5min,用10mL乙酸乙酯清洗均质头,合并提取液4000r/min离心5分钟,上清液经装有10g无水硫酸钠的漏斗脱水于鸡心瓶中,残渣再用20mL乙酸乙酯涡漩洗涤,4000r/min离心5分钟,上清液并入鸡心瓶中,再用10mL乙酸乙酯冲洗漏斗上的残渣合并洗液35℃浓缩至干。[b]净化:用乙酸乙酯2mL×3次漩涡振荡洗涤鸡心瓶,过经5 mL乙酸乙酯活化过的上填1cm高无水硫酸钠的[color=red]硅胶柱([/color][color=red]LC-Si0.5g / 6mL[/color][color=red]硅胶固相萃取小柱)[/color],待洗涤液流完接近硅胶柱中无水硫酸钠顶端时,再用乙酸乙酯洗, [color=red]弃去前[/color][color=red]9 mL[/color][color=red]洗液,[/color]继续用乙酸乙酯洗并收集15mL于15mL刻度玻璃离心管中,35℃水浴氮气吹干,用1mL乙酸乙酯(色谱纯)定容,上机测试。[/b]说明:[align=left]1)提取剂乙酸乙酯极性较强,能有效地将食品中的甲胺磷提取出来,且样品基质中的共提取杂质相对较少;使用乙腈提取时共提取杂质稍多。使用无水硫酸钠一方面配合均质器研磨,增加分散的均匀度,加强溶剂与样品的接触,提高提取效率,另一方面可以将样品中的水分以结晶水的方式除去,既不对甲胺磷产生吸附,又避免甲胺磷溶于水导致回收率的损失。配合超声波辅助提取,进一步提高提取效率。实验时需先加乙酸乙酯后加无水硫酸钠,以免无水硫酸钠结块导致均质困难。由于本实验对水分的残留较为敏感,提取时要尽可能将水分除干净,否则影响刭PSA填料的吸附性能,净化效果变差; 影响到Lc—si柱的吸附性能,可能改变柱上的洗脱规律,甚至导致实验的失败。可将无水硫酸钠在 650℃焙烧约4 h后备用,必要时增加用量。[/align] 作用机理研究: LC—Si柱/乙酸乙酯选择洗脱净化是本前处理方法的核心步骤。Lc-Si柱是经典的正相同相萃取柱,基于正相原理使杂质吸附于柱上,目标化合物随溶剂洗出,一般使用中等偏弱极性的溶剂洗脱。乙酸乙酯是极性较强的溶剂,在这种介质中,大量中强极性及弱极性杂质均难以保留而与目标化合物一起洗出,导致净化步骤失效。本实验正利用了在乙酸乙酯介质中大量杂质均难以保留的特点,[color=red]使其先于甲胺磷流出[/color][color=red]LC[/color][color=red]—[/color][color=red]Si[/color][color=red]柱,然后甲胺磷在特定阶段流出再与仍然吸附于柱上的强极性杂质分离,达到了良好的净化效果。[/color][color=red][/color]此法适用于各种复杂基质中的甲胺磷、乙酰甲胺磷的检测(如果没有经过[color=red]LC[/color][color=red]—[/color][color=red]Si[/color][color=red]柱净化处理且洗脱液的前[/color][color=red]9mL[/color][color=red]洗脱液要弃去,有的样品基质会再甲胺磷、乙酰甲胺磷的出峰位置如茶叶基质中的咖啡因一样出现很大的坡峰,给甲胺磷、乙酰甲胺磷的定量造成干扰[/color])净化处理。结论:综合所述:茶叶中甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷中有机磷的检测要分成两种不同的净化方法进行处理。样品处理(方法一)适用于茶叶中甲胺磷、乙酰甲胺磷、氧乐果的净化处理方法,因为此种净化处理方法会同时萃取出茶叶基质中的咖啡因,茶叶富含咖啡因基质,在[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]中难以消除,会给在咖啡因出峰位置相近的化合物的定量产生干扰(如:毒死蜱、马拉硫磷、杀螟硫磷等)样品处理(方法二)适用于茶叶中毒死蜱、马拉硫磷、杀螟硫磷的测定,如果茶叶样品没有检测甲胺磷、乙酰甲胺磷、氧乐果时,尽量采用此种的净化处理方法,茶叶中的咖啡因用饱和氯化钠水溶液和正己烷液液分配处理后,水溶性杂质、咖啡因及农药(甲胺磷、乙酰甲胺磷、氧乐果、久效磷)会进入饱和氯化钠水溶液层,而其它的农药则留在正己烷层,茶叶样品基质中的咖啡因大谷峰已消失。样品处理(方法三)是遇到个别复杂基质(如有次我们要检测到含茶制品:速溶麦香红茶)时,必要时采用的净化处理方法,过硅胶柱(LC-Si柱)用乙酸乙酯洗脱且弃去前9mL洗脱,收集后面的15mL左右的洗脱液可保证基质中的杂质在前面9mL时洗出弃去,而目标物(甲胺磷、乙酰甲胺磷)被准确定量地收集到。[align=left][/align]
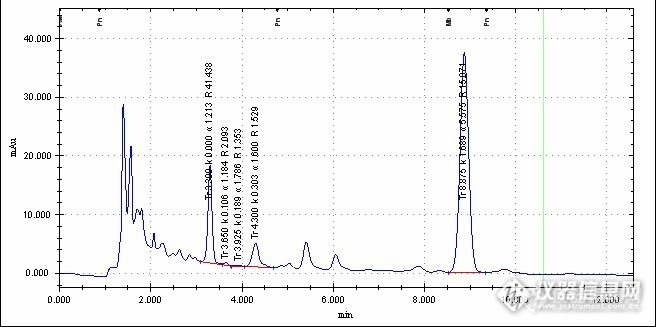
锁阳咖啡中咖啡因含量的测定锁阳咖啡以天然锁阳为基础,配以速溶咖啡,是近几年涌现出的一种新型的固体饮料。锁阳主产于甘肃、青海、内蒙,其中以甘肃嘉酒地区的锁阳产量最大、质地最优。锁阳可调节生理机能、均衡营养、促进血液循环、滋肝健肾。锁阳咖啡是锁阳和咖啡的有机结合,越来越受到消费者的青睐。但是人们在饮用锁阳咖啡的同时却容易忽略咖啡的品质,咖啡因是咖啡中的一种重要成分,也是衡量其质量的一项重要指标,作为一种中枢兴奋剂,能兴奋大脑皮层,但易上瘾,因而国家对其制定了相应的标准。本实验用SN/T 1391-2004 《进出口速溶咖啡检验规程》中速溶咖啡中咖啡因的测定方法测定锁阳咖啡中咖啡因的含量。http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454526_2764104_3.jpg1.方法提要样品用水溶解过滤后,用配有紫外检测器的高效液相色谱(HPLC)测定咖啡因,外标法定量。2.试剂和材料所有试剂除特殊注明外,均为分析纯,水为超纯水。2.1 乙睛:色谱纯;2.2 咖啡因标准品:纯度≥99%;2.3 标准储备液(100m g/L),准确称取 。0.0100 g 咖啡因标准品于100m L容量瓶中,用水溶解并定容至刻度,作为标准储备液。根据需要再用水将标准储备液稀释成适当浓度的标准工作液。3.仪器和设备3.1高效液相色谱仪配紫外检测器。3.2超声波振荡器。4.测定步骤4.1 提取称 取 0.1g(准确至 0.0001 g )均匀试样于 100m L容量瓶中,加人 80m L水,置超声波振荡器中超声20 min,冷却后用水定容至刻度并混匀,过0.45滤膜后,供HPLC测定。4.2 测定4 .2 .1 色谱条件a) 液 相色谱仪,配紫外检测器,检测波长273n m;b) 色 谱柱:C,。柱(25cm×4.6m mID,5um)柱或相当柱;c) 流 动相:乙睛一水一乙酸(16+83十1);d) 流 速 0.5 m L/min;e) 进 样 量 5uL4.2.2 色谱测定根据样液中咖啡因的含量情况,选定峰面积相近的标准工作溶液。标准工作溶液和样液中的咖啡因的响应值均应在仪器的检测线性范围内对标准工作溶液和样液等体积参插进样测定。在上述色谱条件下,咖啡因的保留时间约为9min。 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291536_454528_2764104_3.jpg 标准溶液色谱图 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454529_2764104_3.jpg 标准曲线 http://ng1.17img.cn/bbsfiles/images/2013/07/201307291537_454530_2764104_3.jpg 样品色谱图 4.2.3[/size
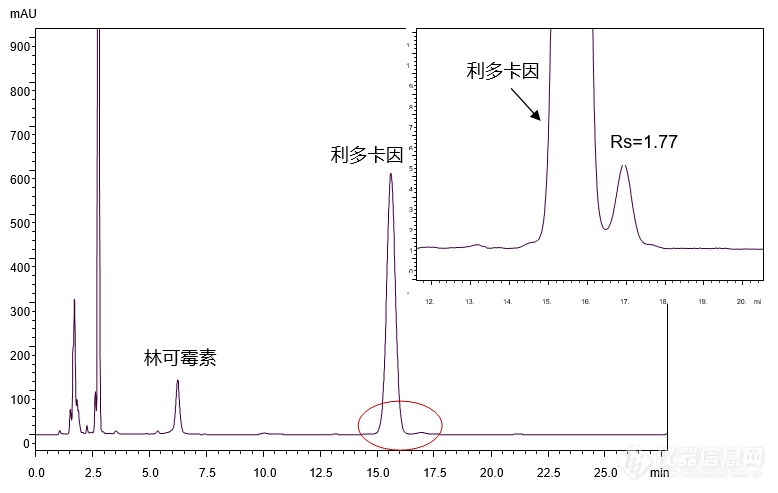
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
ACN 乙腈 Acetonitrile AUFS 满量程的吸光度单位 Absorbance units, full scale As 峰不对称因子 B 二元流动相中的强溶剂;例如:反相HPLC的甲醇/水混合液中的甲醇 BSA 牛血清白蛋白(一种蛋白质) Bovine serum albumin CAF 咖啡因(中性溶质) Caffeine CRF 色谱响应因子 Chromatographic response function;色谱图总分离度的定量指标 dc 色谱柱内径(cm) DMOA 二甲基辛胺 Dimethyloctylamine DNB 2,4-二硝基甲酰(基) 2,4-Dinitrobenzoyl dp 色谱柱填料的粒度(cm) DRYLAB 液相资源公司(LC Resources INC.)的计算机模拟软件。DRYLAB I用于等度预测,DRYLAB G用于梯度预测 F 流动相的流速(ml/min) FC-113 1,1,2-三氟-1,2,2-三氯乙烷 GPC 凝胶渗透色谱法 Gel-permeation chromatography HA 酸性溶质,能电离出A- Hex 己烷 Hexane hr 二相邻谱带之间的谷高 HVA 高香草酸 Homovanillic acid h’ 峰高 h1,h2 相邻谱峰1和谱峰2的峰高 IEC 离子交换色谱法 Ion-exchange chromatography IP 离子对 Ion-pair IPC 离子对色谱法 Ion-pair chromatography J 色谱峰强度参数 K’ 所给谱峰的容量因子,k’=(tR-t0)/t0=tR’/t0,tR=t0(1+k’) k 梯度洗脱过程中,某溶质的k’的平均值或有效值 kw 以水做流动相k’的外推值 k1,k2 相邻谱峰1和谱峰2的容量因子 L 色谱柱长度(cm) Lc 检测器流动池光路的长度(cm) M 溶质的分子量 MC 二氯甲烷 Methylene chloride MDST 混合设计统计技术 Mixture-design statistical technique;一种优化流动相的软件 MeOH 甲醇 Methanol MTBE 甲基叔丁醚 Methyl-t-butyl ether TFA 三弗乙酸MW 溶质的分子量 N 色谱柱塔板数 NAPA N-乙酰普鲁卡因胺 N-Acetylprocainamide(碱性溶质) N0 检测器的基线噪音 ODS 十八烷基硅烷 Octadecylsilyl P 色谱柱的压力降[通常以巴(bar)表示,也用psi;另外,也用作柱极性参数
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)
咖啡因的特性能够提升大脑和身体的能力。1979年,研究人员对比了摄取咖啡因的运动运与未摄取的运动员在长距离自行车项目中的表现,发现摄入咖啡因的运动员表现增加7%。其它也有很多研究证实了咖啡因对人体机能有着短时的提升作用。咖啡因也能与其它药结合提高药效。咖啡因能使头痛药的功效提高40%,并能提高身体吸收药品的速度,缩短药品起作用的时间。所以,在很多药品成分中都会有咖啡因。鉴于咖啡因的特性及作用,人们会对其有一定的依赖性。但由于个体差异,每个人可以摄入的咖啡因的量是不同的,这是由人体基因决定的。对于想知道自己一次摄入多少咖啡因才是最合适的人来说,要不赶紧做个咖啡因代谢基因检测了解一下~有咖啡因成分的咖啡、茶、软饮料及能量饮料十分畅销各类饮品中咖啡因含量对比表【咖啡】 咖啡因含量 80mg(一般含量范围 40-170mg)【红茶】 咖啡因含量 40mg(一般含量范围 25-110mg)【乌龙】 咖啡因含量 30mg(一般含量范围 12-55mg)【绿茶】 咖啡因含量 20mg(一般含量范围 8-30mg)【普洱】 咖啡因含量 2mg (含量极低)【麦茶】 咖啡因含量 0mg【可乐】 咖啡因含量 50mg(一般含量范围 40-80mg)【红牛】 咖啡因含量 80mg(一般含量范围 80-100mg)







 我要推广仪器
我要推广仪器
 下载APP
下载APP