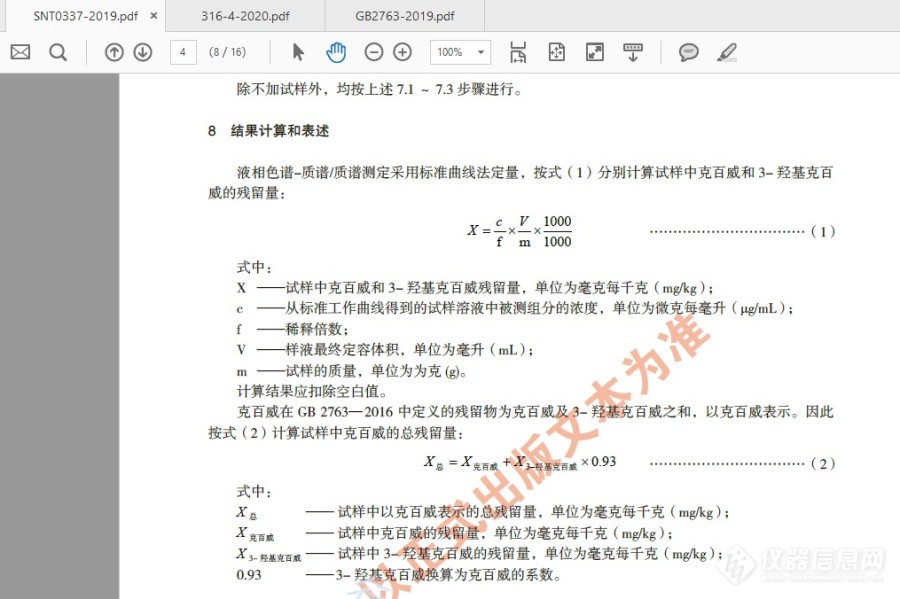
关于克百威(以克百威和3-羟基克百威之和计)结果计算问题的请教。想请教一下:当样品中克百威,3羟基克百威都有检出的情况下,按照GB 2763-2019的要求,克百威是“以克百威和3-羟基克百威之和计”,请问是直接将两者进行求和,还是进行换算后再求和。例如:某蔬菜样品中克百威检出值为 A mg/Kg,3-羟基克百威检出值为 B mg/Kg,现在要填报“克百威(以克百威和3-羟基克百威之和计)”的结果,应该怎样计算才正确。计算方式一:样品中 克百威(以克百威和3-羟基克百威之和计)含量=(A+B)mg/Kg,计算方式一:参照 SN/T 0337-2019标准中的计算方式,样品中 克百威(以克百威和3-羟基克百威之和计)含量=(A+B*0.93)mg/Kg。请各位老师指教。附:SN/T 0337-2019 部分截图:[img=,690,459]https://ng1.17img.cn/bbsfiles/images/2021/04/202104301427586649_3321_3463672_3.jpg!w690x459.jpg[/img][img=,690,459]https://ng1.17img.cn/bbsfiles/images/2021/04/202104301427586649_3321_3463672_3.jpg!w690x459.jpg[/img]
2763 中克百威和3羟基克百威都用克百威表示!这样还有必要在资质认定的时候把3羟基克百威当参数来认证吗?
今年我单位做丁硫克百威残留登记试验,不知道有没有试验单位有3-羟基克百威和3-乙基克百威,如能提供少许(几十毫克就行),真的不胜感激,联系人:万宇 0551-5854938 /0551-5851928 /13866151614
用761方法液相柱后衍生法做涕灭为亚砜、涕灭威砜和三羟基克百威都没有出峰,不知道在前处理的过程中应该注意什么才能出峰?求高手赐教!我们要计量认证考核,急!
各位老师,克百威(克百威和三羟基克百威)检出限怎么写,两者之和么么
请教各位大侠,叔丁基羟基茴香醚(BHA), 2,6—二叔丁基对甲酚(BHT), 敌百虫可采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析吗?
用乙醚萃取丹霞茶叶烘干叶,直接过滤膜用于GC-MS检测,发现茶叶中二丁基羟基甲苯相对含量达百分之三十多,这种现象是正常的吗?二丁基羟基甲苯不是对人体有害的吗,茶叶中含量怎么如此高呢?
如何能使羧酸上羟基的氢原子显示出来?谢谢!
测样时经常会遇到带有羟基的化合物,如4-羟基香豆素的衍生物等,请问各位楼主,有什么办法可以尽量使羟基峰显示出来?
你们进液质的涕灭威亚砜,涕灭威砜3-羟基克百威标样从哪里买?什么规格型号,我们买的国标液是钠盐形式。好友回复:你到国家标准物质网查询, GSB和SB是农业部环境保护科研监测所制的,BW是国家计量院的,CDCT是德国Dr的,一般是固体的
我们是用水杨酸捕捉羟基自由基,以前的做法是用C18的色谱柱,5%的乙醇做流动相,流速是0.3,把标准样用35%乙醇稀释400倍进样,以前的标准样是可以检测出两个峰的(水杨酸和2,5-二羟基苯甲酸),但后来柱子坏了换了一根色谱柱,也是C18的,但不同型号的,用回原来的方法就无法出峰,只有一个溶剂峰,但两种主要物质出不来,后来把流动相换成10%的乙醇、30mMol醋酸钠醋酸缓冲液(ph4.9)都试过还是无法出峰。后来老师让我们用样品原液不稀释进样,峰是出来了,但峰高度很低而且面积也不大,而且很多杂质峰,峰型也很奇怪,重点是水杨酸以及2.5二羟基苯甲酸的保留时间几乎一样,无法分离。以前是标准样稀释400倍就能出峰的,现在为什么用原液峰面积也这么小?还有保留时间一样应该怎么处理?很急啊,谢谢。
用DMSO溶剂做黄酮类化合物的氢谱时,酚羟基的信号有时能够做出了,有时做不出来,是不是羟基氢被置换的原因?是如何置换的?
在做二羟基丙酮的硅烷化分析的时候,出现一个问题:当标准样品的量比较少的时候比如10mg左右的时候,气相上出来的是二羟基丙酮的硅烷化的峰,但是当标品的来那个超过20mg的时候会出来二羟基丙酮硅烷化峰以及二羟基丙酮二聚体硅烷化峰,不知哪位专家做过此物质的分析,可否指点一下。
[img]http://ng1.17img.cn/bbsfiles/images/2006/01/200601121509_12983_1678476_3.gif[/img] 纳米氧化硅是经过硅烷偶联剂改性过的。偶联剂中含有-NH2,-CH2,-CH3O。不知从图中能否判断出:偶联剂已经键合到氧化硅粒子表面了 ?氧化硅表面的羟基在红外谱图上能表现出来吗? 真心求教,请大家帮忙 :)
测试水体中的COD值,氧化方式有重铬酸钾,高锰酸盐,羟基自由基。做成在线的仪器的就分别有CODcr,CODmn,另外紫外光谱扫描的CODuv。而且这几类的厂家及产品很多。使用羟基自由基方式的做成在线仪器的主要是德国LAR,最近发现国内出现了一款使用羟基自由基来测试COD的便携式仪器IGS 20。由广州盈思传感科技有限公司研发生产。其与CODuv相同,都不需要使用试剂,不会造成二次污染,不同的是他还是属于氧化法。 大家觉得羟基自由基氧化法测试COD是否可行,或者说是否能被接受呢?
请问有做1,5-二羟基萘氧化为5-羟基-1,4-萘醌(胡桃醌)的吗?怎么计算反应的转换率啊?用液相还是[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]更好一些呢,我用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]没测出来,柱温为280度。谁有经验哪?麻烦指导一下。拜托了
8-羟基喹啉重量法(GB/T 5059.1-1985)1.方法提要试样以硝酸-氯酸钾饱和溶解分解,钼成钼酸析出,用EDTA络合铁等杂质,加氨水溶解钼酸按,过滤。取部分溶液,pH约4.8醋酸-醋酸按缓冲溶液中,以8-羟基喹啉沉淀钼用玻璃坩埚过滤,在130~140℃电热干燥箱内干燥称重,借此测得钼的含量。本方法适用于钼精矿中钼量的测定。测定范围:大于10%2.试剂氨水(比重0.9)硝酸-氯酸钾饱和溶液:加氯酸钾在硝酸(比重1.42)中,成饱和溶液。乙二胺四乙酸二钠ETDA(C10H12FeN2NaO8• 3H2O)(5 %)。甲基橙(0.1%)。盐酸(1+1)。醋酸-醋酸铵缓冲溶液(pH4.8):280克醋酸铵,用水溶解后,加220毫升冰醋酸,再用水稀释至1000毫升,混匀。8-羟基喹啉(3%):30克8-羟基喹啉溶解在1000毫升2mol/L醋酸中(冰醋酸118毫升)。3.分析步骤称取0.2305克试样,置于250毫升烧杯中,加15~20毫升硝酸-氯酸钾饱和溶液,待剧烈作用后,低温加热分解并蒸发至溶液约剩5毫升,取下,冷却。用少量水吹洗表面皿及烧杯内壁,加20毫升5 % ETDA溶液,摇匀,用氨水(比重0.9)中和至钼酸沉淀全部溶解并过量2毫升,冷却至室温,移入100毫升容量瓶中,用慢速滤纸干过滤,移取50.00毫升试液,置于250毫升烧杯中,加1滴0.1%甲基橙指示剂,用盐酸(1+1)中和至溶液颜色刚好变红,加5毫升醋酸-醋酸按缓冲溶液,用水稀释至溶液约120毫升,煮沸,取下。在不断搅拌下,徐徐加10毫升3%8-羟基喹啉溶液,置于低温电炉上静置2~3分钟。取下,用已在130~140℃干燥过并称重的3~4#玻璃坩埚过滤,用热水洗涤烧杯3~4次,洗涤沉淀8~10次,将坩埚连同沉淀置于130~140℃电电热干燥箱中干燥1小时,取出,置于干燥器内冷却至室温,称重。并反复千燥至恒重。钼的百分含量按下式计算; 式中;W1——玻璃坩埚与8-羟基喹啉钼的重量(克);,W2——玻璃坩埚的重量(克);V——试液总体积(毫升);V1——分取试液体积(毫升);W——称样量(克);0.2305——8-羟基喹啉钼换算成钼的系数。4.允许差含钼量(%)允许差(%)≤200.2520.01~3000.3030.000.405.注意事项①加硝酸-氯酸钾饱和溶液时应防止加入固体氯酸钾。如试样分解不完全应重复加硝酸-氯酸钾和溶液至试样分解完全。②此时不宜将溶液煮沸,否则沉淀会溅跳粘附在烧杯内壁上不易洗净。
石油类的测定HJ637-2012中规定当样品中明显含有羰基和羟基时要在报告中说明,这个明显含有羰基和羟基是怎么确定的。。我们这有一个地表水,在3030和2960中间出了一个峰。我怎么知道这个水里面有羰基和羟基呢???是通过谱图能看出来吗?
主要是测定羟基的伸缩振动引起的红外吸收,吸收峰在3000-4000cm-1之间,这个波段恰好是空气中水分吸收干扰比较大的区域,而且有些材料中羟基的含量很低,10到几百ppm之前用过nicolet的5700和6700,附加了氮气吹扫,但效果仍然不是非常理想,而且太费氮气最近想买一台红外,附带显微镜的,哪位推荐一款,对3000-4000cm-1之间测定效果比较好,背景噪音相对低的。想用nicolet的,主要是软件简单,其他人来用的时候也容易上手,有哪款可以推荐的?多谢各位了补充一下:主要是通过附带的显微镜微区分析,测定的对象是固体,主要是天然的硅酸盐矿物--双面抛光的玻片
想用薄层色谱法验证是否合成了4-羟基吡啶,试了丙酮和环己烷作为展开剂,均没有好的效果,想请问下应该用何试剂呢?顺便问一下,如果是固体样品要点样,应用什么试剂来溶解呢。
GB5009.31-2016对羟基苯甲酸 这个国标中是用乙醚提取酱油中的对羟基苯甲酸甲酯但是如果酱油中添加的是对羟基苯甲酸甲酯的盐,也就是对羟基苯甲酸甲酯钠 ,是不是就很难把这个钠盐提取出来呢?[img]https://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img] 问题有点多,我都不好意思问了。但是不搞明白,心里不舒服
5-羟基癸酸乙酯是由delta-癸内酯制成的,但打出来的图尾段有一个较大的峰,是放久后产生,还是反应不好生成的呢?查过资料正常的图谱是没有这个东西出来的,请大家求解一下。
购买克百威的标液,除了买克百威,还要买三羟基克百威,那么丁硫克百威和他们之间的关系是什么呢?
诚心请教2,5-二羟基-1,4-二噻烷气相色谱分析的条件...在线跪等中...........来位高人指点一二吧
有做过8-羟基喹啉或8-羟基喹啉硫酸盐的液相实验吗?为什么分不开啊,用的是化妆品检疫的方法,求助!求助!
我们用水杨酸浸渍膜来捕获羟基自由基,捕获液尝试过用0.05水杨酸溶于500ml 35%乙醇(这是过去师姐总结的方法),但我们现在用这个方法无法检测出2.5-二羟基苯甲酸,只检测出水杨酸,标准样是可以出峰的,所以色谱仪和色谱柱应该是没有问题,那么方法应该怎么改进。问题来了:水杨酸捕获液的浓度应该多少?具体怎么配制?检测波长设定多少?捕获时间要多长?
药典规定:对羟基苯甲酸乙酯(也就是尼泊金乙酯),用电位滴定法滴定到第2个终点。我用ZDJ-3D全自动电位滴定仪,滴定等当点2:得出的结果的 百分之200多,用公式:vFT/m 可推回,且导数曲线有两个峰。(以去空白)不知道是电位滴定仪的设置方法是问题,还是第二个滴定终点在计算上有所不同。如有高手做过这种以第2个滴定终点为终点的含量测定,请指教。谢谢
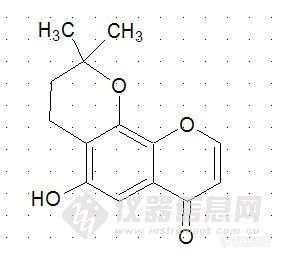
http://ng1.17img.cn/bbsfiles/images/2011/02/201102240945_279159_2238889_3.jpg这个支链 可以和8位羟基成环(图就是和8位成环后) 也可以和 6位羟基成环 但是怎么去认定具体是和那个成环呢? 环上和氧相连的碳没有氢 那么做HMBC 就不可能和母核出相关峰, 紧靠碳谱的化学位移能说明问题吗?还是有其他的什么办法?谢谢各位浏览!
求助:对羟基苯甲酸丁酯钠和对羟基苯甲酸丙酯钠的相关行业标准,国内、国外的标准都可以。
糖的几个羟基化学环境相似,核磁图如何确定哪个羟基发生了反应







 我要推广仪器
我要推广仪器
 下载APP
下载APP