请问,甲基酚—麝香草酚蓝试剂遇见碱应该显什么色?我做的实验怎么是紫色的!
跪求,甲基磺草酮的使用方法及液相分析方法
草甘膦、氨甲基膦酸和草甘膦内标衍生: 取1ml 20g/L上述物质标准溶液(水作溶剂),分别加0.5ml 50g/L硼酸钠、0.5ml 10g/LFMOC-Cl衍生剂,37℃衍生2h。SN/T 1923是缓冲盐和衍生剂各加0.2ml,并且衍生剂浓度是1g/L,担心衍生不完全我多加了,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS上扫描啥也没有或者说响应极低不稳定,倒是有几个离子响应很稳,比如草甘膦扫出来是179,365和397,正常来说179、392。 后面,我又直接拿草甘膦和氨甲基膦酸(未经衍生)上机扫描,文献上有负离子扫描出峰的,我试了一下,草甘膦(分子量169)扫出来是168,响应较低不稳定,但合理。就是说仪器没问题,标准品没问题。 问题来了,到底是哪里出了问题?衍生剂我都是临配现用的,顶多也是过量了而已
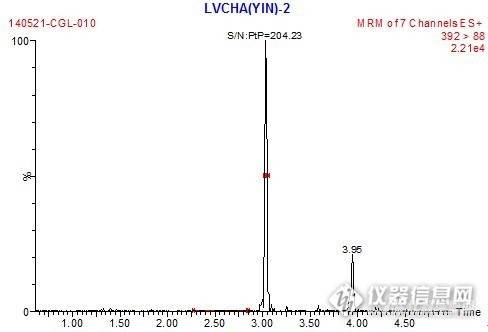
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
自来水7月1日实行新国标,特把自来水中草甘膦、胺甲基硫酸的检测方法拿来共同分享
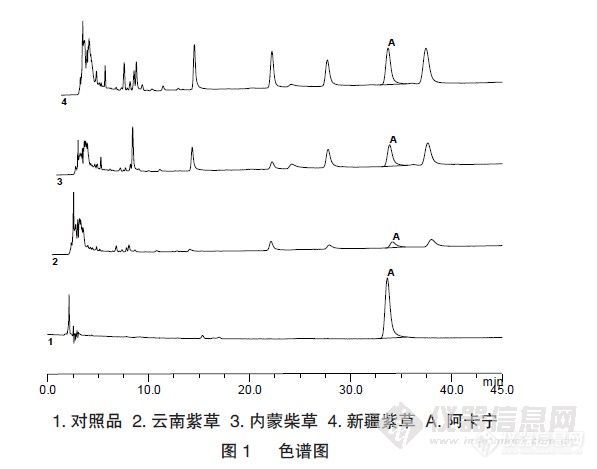
【作者】 谢清春; 陈燕忠; 吕竹芬;【Author】 XIE Qingchun CHEN Yanzhong LV ZhufenInstitute of Material Medica, Guangdong Pharmaceutical College, Guangzhou 510006, Chin【机构】 广东药学院药物研究所;【摘要】 目的测定不同产地的紫草中β,β’-二甲基丙烯酰阿卡宁(以下简称阿卡宁)的含量,比较不同溶剂对紫草中阿卡宁的提取率,并对阿卡宁的乙醇溶液稳定性进行考察。方法采用高效液相色谱法测定阿卡宁的含量,色谱柱为Dikma Diamonsil C18柱(200mm×4.6mm,5μm),以乙腈-水-甲酸(700:300:0.5)为流动相,流速1mL·min-1,检测波长275nm。测定了内蒙紫草、新疆紫草和云南紫草中阿卡宁的含量,考察了75%乙醇、乙醇、石油醚和液体石蜡对紫草中阿卡宁的提取率;将阿卡宁0.1mg·mL-1乙醇溶液于冰箱4℃存放,定期进样测定,并与新配制时测定的峰面积比较。结果测定的三种紫草中,以新疆紫草含量最高;乙醇和石油醚对阿卡宁的提取率高而75%乙醇和液体石蜡的提取率低;阿卡宁的乙醇溶液随存放时间的增加而峰面积变小。结论提取溶剂对紫草中阿卡宁提取率影响较大;由于阿卡宁溶液的不稳定性,选择阿卡宁作为评价含紫草药材制剂的指标成分并不合适。 更多还原【Abstract】 Objective To determine the β,β’-dimethylacrylalkannin in lithospermum erythrorhizon, compare the extractingrate of different solvents to β,β’-dimethylacrylalkannin and study the stability of standard β,β’-dimethylacrylalkanninsolution of alcohol. Methods A HPLC method was used to determine the β,β’-dimethylacrylalkannin. Dikma Diamonsil C18chromatogram column(200 mm×4.6 mm, 5μm) was used, Acetonitrile-water-Formic acid(700:300:0.5) as mobile phase, flowrate at 1mL·min-1, detection wavelength at ... 更多还原【关键词】 紫草; β,β’-二甲基丙烯酰阿卡宁; 提取率; 高效液相色语法; 稳定性; 【Key words】 Lithospermum erythrorhizon; β,β’- dimethylacrylalkannin; Extracting rate; HPLC; Stability; http://ng1.17img.cn/bbsfiles/images/2012/08/201208201119_384576_2352694_3.jpg
哪里有卖N,N-二甲基对苯二胺盐酸盐或者是草酸盐的,价格多少?

“超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留”是本人去年开展大豆中草甘膦检测项目整个试验过程的总结,欢迎各位老师和同行批评指正,该文章还未在任何刊物上发表。[align=center][b]超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留[/b][/align][align=center][/align][align=center]户江涛[/align][align=center](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了快速检测大豆中草甘膦和氨甲基膦酸残留量的分析方法。试样经水超声提取,二氯甲烷去除脂肪,C[sub]18[/sub]固相萃取柱净化后,在硼酸钠缓冲溶液中与9-芴甲氧羰酰氯(FMOC-Cl)进行衍生反应,其衍生产物在C[sub]18[/sub]色谱柱上以 2 mmol/L 乙酸铵溶液和乙腈为流动相,进行液相色谱分离:质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,草甘膦和氨甲基膦酸在0.001~0.5 mg/L范围内线性关系良好,相关系数(R)分别为0.9996和0.9993,定量限(LOQ)均为0.01mg/kg。在空白大豆样品添加浓度为0.02、0.2、2 mg/kg 时,草甘膦和氨甲基膦酸的平均回收率分别为80.2%~91.5%和77.7%~89.3%,相对标准偏差(RSD)分别为3.37%~6.96%和4.11%~8.27%(n=6)。本方法快速、简便、灵敏,适用于大豆中草甘膦和氨甲基膦酸残留的同时检测。关键词:超高效液相色谱-串联质谱;大豆;草甘膦;氨甲基膦酸;衍生反应[align=center]Determination of glyphosate and its metabolite aminomethyl-phosphonic acid residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Testing and Analysis Center of Heilongjiang Academy of Land Reclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A method[b] [/b]was developed for the determination of glyphosate(PMG) and aminomethyl-phosphonic acid(APMA) residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). After extracted with water under ultrasonication, the sample was defatted with dichloromethane and purified by C[sub]18 [/sub]solid phase extraction cartridge, and then PMG and APMA were derivatized using 9-fluorenylmethoxycarbonyl(FMOC-Cl) in borate buffer for 2 h.The derivatives of PMG and APMA were separated on a Waters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of 2 mmol/L ammonium acetate and acetonitrile, and finally detected by positive eletrospray ionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reaction monitoring(MRM) mode.The results showed the linearities of PMG and APMA were good in the concentration range of 0.001~0.5 mg/L ,and the correlation coefficients were 0.9996 and 0.9993 respectively. The limit of quantification(LOQ) of PMG and APMA was both 0.01mg/kg. At the spiked levels of 0.02、0.2、2 mg/kg in the blank soybean samples, the mean recoveries of PMG and APMA were 80.2%~91.5% and 77.7%~89.3% respectively, and the relative standard deviation(RSD) of PMG and APMA were 3.37%~6.96% and 4.11%~8.27% res-pectively(n=6).This method is fast,simple,sensitive, and suitable for simultaneous determination of PMG and APMA in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) soybean glyphosate(PMG) aminomethyl-phosphonic acid(APMA) derivatization草甘膦(Glyphosate,PMG)又名镇草宁、农达,分子式为C[sub]3[/sub]H[sub]8[/sub]NO[sub]5[/sub]P,是1971年美国孟山都公司研发的一种有机磷除草剂,因其兼具内吸、传导性、灭生性及非选择性,同时不易在生物体内累积,故广泛应用于农业生产中一年生和多年生杂草防除,是目前世界上应用最广、生产量最大的除草剂[sup][/sup]。草甘膦及其在植物中的主要代谢物氨甲基膦酸(Aminomethyl-phosphonic acid,APMA,分子式为CH[sub]6[/sub]NO[sub]3[/sub]P)均属于强极性、易溶于水的高沸点化合物,具有不易挥发、无紫外吸收等特性,因此用常规方法分析检测十分困难[sup][/sup]。 目前, PMG和APMA残留检测的方法主要有色谱法(GC[sup][/sup]、LC[sup][/sup]、IC[sup][/sup])、质谱法(GC/MS[sup][/sup]、ICP/MS[sup][/sup]、LC/MS/MS[sup][/sup])、光谱法[sup] [/sup]等。光谱法虽然操作简便,但其灵敏度不高,而[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法[sup][/sup]只能适用于水样等简单基质,用于植物源样品检测时干扰太大;用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]技术检测时,需要将PMG和APMA衍生转化为可气化物质,其引入试剂多、过程相对繁琐,效率较低[sup][/sup];用LC/MS/MS法直接检测时[sup][/sup],由于PMG和APMA分子量(分别为169、111)均较小,其主要碎片离子的质荷比多在100以下,检测实际样品时受基质干扰严重,灵敏度较低,因此柱前衍生——[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]法成为近年来国内外检测PMG和APMA残留的主流方法[sup][/sup]。以9-芴甲氧羰酰氯(FMOC-Cl)做为衍生试剂,在硼酸盐缓冲溶液中与PMG和APMA水提取液相容性好,过程简单,其衍生产物在LC/MS/MS中响应信号高,碎片离子干扰小,适合定性定量分析。 目前,采用柱前衍生——LC/MS/MS法检测茶叶、稻米等基质中PMG和APMA残留的报道很多[sup][/sup],专门针对大豆基质的报道很少。行业标准[sup][/sup]的适用范围虽然包括了大豆基质,但该方法在实验过程中试剂用量大、操作繁琐(反复调pH值)、衍生时间长(需过夜),尤其是使用阳离子交换柱(CAX)洗脱时需要加入11 mL 1%的盐酸甲醇水(20/80,v/v),水分含量过高导致旋转蒸发时很难蒸干,容易造成PMG和APMA回收率不稳定。本文专门针对大豆这类高蛋白、高脂肪含量的特殊基质,采用纯水作为提取试剂,二氯甲烷去除脂溶性杂质,C[sub]18[/sub]固相萃取小柱净化后采用FMOC-Cl衍生,最后用UPLC/MS/MS测定。该方法前处理过程简便、快速、灵敏度高,适用于大豆中PMG和APMA的残留检测。[b]1 实验部分[/b]1.1 材料与试剂 草甘膦、氨甲基膦酸(纯度≥99%,德国Dr.Ehrensorfer公司);FMOC-Cl(纯度99%,Sigma公司),使用时配置成10g/L的丙酮溶液;乙腈、二氯甲烷、甲酸、乙酸铵(色谱纯,美国Fisher公司);十水四硼酸钠(优级纯,天津市科密欧化学试剂有限公司),使用时配置成50g/L的水溶液;实验用水为Millipore纯水仪制备;C[sub]18[/sub]固相萃取小柱(200mg/3ml,美国Agilent公司)。1.2 仪器与设备 Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);CR21GⅢ型高速离心机(HITACHI公司);KQ5200DB型台式超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司)。1.3 标准溶液的配置 分别称取草甘膦和氨甲基膦酸标准品10mg(精确到0.1mg),用水溶解并定容至10mL,配置成质量浓度为1.0 mg/mL标准储备液,于4℃冰箱保存待用;使用时用水逐级稀释成所需浓度的混合标准工作液。1.4 样品前处理 提取:称取粉碎均匀后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0mL超纯水,涡旋混合30 s并超声提取20 min后,以10000 r/min离心3 min,将上清液转移至另一离心管中,加入5 mL二氯甲烷涡旋混合30 s,以10000 r/min离心3 min,上清液待净化。 净化:取2.5 mL上清液加入到C[sub]18[/sub]固相萃取柱(使用前依次用3mL甲醇和3mL超纯水活化)中,弃去最初的几滴流出液(约0.5 mL),将剩余部分用5 mL玻璃管收集,待衍生。 衍生:取1.0 mL净化液于5 mL离心管中,依次加入1.0 mL 50g/L的硼酸钠溶液和 1.0 mL 10g/L的FMOC-Cl衍生液,混匀后室温下衍生2 h,以10000 r/min离心3 min,取上清液过0.22 mm有机系微孔滤膜后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件 液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:2 μL;流动相A:乙腈;流动相B: 2 mmol /L 的乙酸铵水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。 质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1 PMG-FMOC和 AMPA-FMOC的MRM质谱参数[/align][align=center]Table 1 MRM parameters of PMG-FMOC and AMPA-FMOC[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Cone/V[/align][/td][td][align=center]Parent ion/(m/z)[/align][/td][td][align=center]Daughter ion/(m/z) [/align][/td][td][align=center]Collision energy/V[/align][/td][/tr][tr][td][align=center]PMG-FMOC[/align][align=center] [/align][align=center]AMPA-FMOC[/align][/td][td][align=center]30[/align][align=center] [/align][align=center]30[/align][/td][td][align=center]392[/align][align=center][sup] [/sup][/align][align=center]334[/align][align=center][sup] [/sup][/align][/td][td][align=center]88[/align][align=center]214﹡[/align][align=center]112﹡[/align][align=center]179[/align][/td][td][align=center]14[/align][align=center]8[/align][align=center]11[/align][align=center]20[/align][/td][/tr][/table]﹡quantitative ion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化 流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(乙酸铵水溶液)分别于甲醇、乙腈的流动相体系组合,结果发现两种分析物在酸性体系中分离效果欠佳,峰形拖尾严重,而在非酸性体系中其色谱分离效果得到明显改善,峰形对称;乙腈比甲醇体系洗脱能力更强,可以有效缩短分析时间。故本研究采用乙酸铵水溶液+乙腈流动相体系,并比较了1、2、5 mmol/L三种乙酸铵浓度与乙腈的组合,结果发现随着乙酸铵浓度的增加,目标物响应值虽略有提高但相差不大,而同时仪器背景值却显著升高,综合考虑目标物信号强度、信噪比、色谱分离效果以及分析时间等因素,本实验最终选择了2 mmol /L 乙酸铵水溶液+乙腈分析体系。质谱的选择:PMG、 AMPA对应的衍生物PMG-FMOC、AMPA-FMOC分子量分别为391、333。用超纯水配置10 mg/L 混合标准溶液直接注射到质谱中,在正负离子模式下分别进行母离子全扫描,发现正离子模式下392、334具有很好的响应,然后分别以392、334为母离子进行子离子全扫描,各得到两组丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1。2.2 前处理条件的优化 提取溶液的选择:PMG和APMA属于强极性物质,易溶于水,难溶于有机溶剂,故一般采用极性溶剂提取,如纯水及KOH、NaHCO[sub]3[/sub]溶液等[sup][/sup]。实验发现,用碱性溶液提取后,大豆中脂肪、蛋白等物质会与碱性物质发生反应,导致离心后的提取液异常浑浊,不利于后期净化和衍生,因此本实验采用纯水作为提取试剂,再经二氯甲烷液液萃取去除脂溶性杂质。 净化柱的选择:研究发现,对提取后的溶液不经SPE净化直接进行衍生, PMG和APMA的回收率均不足30%,且精密度很差,这可能是由于大豆中富含脂肪、蛋白质等物质干扰衍生过程,故本实验比较了对脂肪、蛋白质有很好去除效果的C[sub]18[/sub]、中性Al[sub]2[/sub]O[sub]3[/sub]、HLB固相萃取SPE柱的净化效果,结果发现提取液经中性Al[sub]2[/sub]O[sub]3[/sub]净化后,PMG和APMA几乎检测不到;而C[sub]18[/sub]净化后目标物回收率为92.7%、90.8%,HLB为83.6%、80.5%。故本实验选取了净化效果更好,成本相对低廉的C[sub]18[/sub]固相萃取小柱。 衍生条件的优化:FMOC-Cl的衍生机制是在碱性环境下(pH=9.0)通过FMOC-Cl基团取代目标化合物氮原子上的氢,从而生成较稳定的化合物FMOC-Cl。参照行业标准[sup][/sup]及文献报道[sup][/sup]所选用的缓冲液浓度,本实验采用50g/L的硼酸钠水溶液缓冲液体系,设置的衍生试剂质量浓度为1、2、5、10、20 g/L FMOC-Cl丙酮溶液,按照本文1.4步骤对PMG和APMA质量浓度为0.5 mg/L的纯水溶液和大豆空白基质溶液分别进行衍生,结果见图1。结果表明,在纯水溶液中,FMOC-Cl浓度为2 g/L时,PMG和APMA的峰面积已达到最大,随着衍生化试剂浓度的升高,其峰面积无明显变化;而在大豆空白基质溶液中,FMOC-Cl低浓度(1、2g/L)时,PMG和APMA几乎检测不到,其峰面积随衍生化试剂浓度增加而加大,浓度到达一定程度(10 g/L)时,峰面积不再变化。产生这种现象的原因,可能是由于尽管大豆提取液经过了二氯甲烷和C[sub]18[/sub]小柱的净化,但还是会有少量水溶性蛋白、脂肪等杂质残留在净化液中,这些杂质可能会与衍生试剂反应,影响目标物的衍生效果。研究还发现,当FMOC-Cl浓度为20 g/L时,得到的PMG和APMA色谱峰产生拖尾现象,可能是由于衍生试剂化学性质较活泼,其用量大时,过量的FMOC-Cl会迅速转化成FMOC-OH,干扰目标物峰形。在50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液条件下,考察不同时间(0.5h、1h、2h、4h、8h和16h)对衍生效果的影响,结果发现,2 h后PMG和APMA的测定值无明显增加。因此,本实验最终选定的衍生条件为50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液,室温下衍生2 h。[img=,596,378]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_01_2984502_3.png[/img][img=,690,530]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_02_2984502_3.png[/img]2.3 基质效应的考察 基质效应(主要是抑制)是LC/MS/MS仪器检测时经常遇到的现象。由于本实验采用极性很强的水作为提取剂,大豆中的色素、脂肪酸等极性较强的物质也有少部分进入到最后的上机液中,在离子化带电过程中会与目标物产生竞争,抑制目标物的离子化效率。实验考察了用PMG和APMA的纯水标样去标定经过本文1.4步骤处理后的大豆空白基质溶液配置的同浓度标样,其色谱图见图2。结果发现,PMG在纯水和大豆空白基质中峰面积基本一致,而APMA在大豆空白基质中的峰面积仅为纯水中的55.7%,产生了明显的基质抑制效应。为了消除基质干扰,本实验选用大豆样品空白基质配置不同浓度的标准溶液来绘制标准曲线进行校准。2.4 线性范围和定量限 用大豆空白基质溶液分别配置0.001、0.005、0.01、0.05、0.1、0.2、0.5 mg/L的PMG和APMA混合标准溶液,按本文1.4步骤衍生后测定,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,这两种物质在0.001~0.5 mg/L浓度范围内线性良好,相关系数R分别为0.9996和0.9993。以10倍信噪比(S/N)计算,该方法PMG和APMA的定量限(LOQ)均为0.01 mg/kg。[align=center]表2 PMG和APMA大豆基质标准溶液的线性方程、相关系数和定量限(LOQ)[/align][align=center]Table 2 Linear equations,correlation and LOQ of PMG and APMA in the soybean matrix standard solutions[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Linear range/(mg/L)[/align][/td][td][align=center]Linear equation[/align][/td][td][align=center]R[/align][/td][td][align=center]LOQ/(mg/ kg )[/align][/td][/tr][tr][td][align=center]PMG[/align][align=center]AMPA[/align][/td][td][align=center][sup]0.001~0.5[/sup][/align][align=center][sup]0.001~0.5[/sup][/align][/td][td][align=center]Y=889809x+1671.3[/align][align=center]Y=476982x+1161.9[/align][/td][td][align=center]0.9996[/align][align=center]0.9993[/align][/td][td][align=center]0.01[/align][align=center]0.01[/align][/td][/tr][/table]2.5 回收率和精密度 称取大豆空白试样1.0 g,分别添加0.02、0.2、2 mg/kg水平的PMG和APMA混合标样,每个水平重复6次,按照本文1.4步骤前处理方法处理后上机检测,实验结果见表3。从表3可以看出,PMG的平均回收率为80.2%~91.5%,相对标准偏差(RSD,n=6)为3.37%~6.96%;APMA的平均回收率和RSD分别为77.7%~89.3%和4.11%~8.27%。[align=center]表3 大豆中PMG和APMA的加标回收率和相对标准偏差(n=6)[/align][align=center]Table 3 Recoveries and relative standard deviations(RSD)of PMG and APMA spiked in the soybean(n=6) [/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Spiked level(mg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td]PMGAMPA[/td][td][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][/td][td][align=center]80.2[/align][align=center]91.5[/align][align=center]86.8[/align][align=center]77.7[/align][align=center]89.3[/align][align=center]85.9[/align][/td][td][align=center]6.96[/align][align=center]3.37[/align][align=center]3.95[/align][align=center]8.27[/align][align=center]4.25[/align][align=center]4.11[/align][/td][/tr][/table][b]3 结语[/b] 本文建立了超高效液相色谱-串联质谱法(UPLC/MS/MS)测定大豆中草甘膦及其代谢物氨甲基膦酸残留的分析方法。该方法灵敏度高,PMG和APMA定量限(LOQ)达到0.01 mg/kg,能满足大豆产品相关限量标准要求。同时该方法具有较高的准确度和精密度,前处理步骤简单快速,特别适合大批量大豆样品的检测。

气相色谱法对维生素E的原料三甲基氢醌的检测摘要 三甲基氢醌即2,3,5-三甲基氢醌,又名2,3,5-三甲基对苯二酚,是生产维生素E的中间体,其主要用途是用作生产维生素E的主要原料。目前,维生素E已成为国际市场上用途广泛、产销量极大的主要维生素品种,国内外市场前景广阔。目前全国生产能力不能满足国内市场供应不足,部分依赖进口。因此对三甲基氢醌的需求日益增加。而对于三甲基氢醌检测目前国家和行业都没有一个统一的检测标准。为此南京科捷分析仪器应用研究所根据客户的要求应用GC5890C气相色谱仪对2,3,5-三甲基氢醌进行方法研究。实验结果表明:本方法简便,分析速度快。能满足生产质量控制的要求,从而降价低生产成本。关键词 2.3.5- 三甲基氢醌 2,3,5-三甲基对苯二酚 维生素E中间体 气相色谱法一.2.3.5三甲基氢醌气相色谱图 http://ng1.17img.cn/bbsfiles/images/2011/06/201106171053_300271_2242538_3.jpg三、仪器配置 检测项目2,3,5-三甲基氢醌及其杂质色谱仪器型号GC5890C型色谱仪 配有FID检测器毛细管色谱柱0.32*30*0.25专用柱色谱工作站N2000(电脑1台自备)氮氢空发生器 HGT300E 1台或高纯氮、氢气、空气钢瓶各一瓶
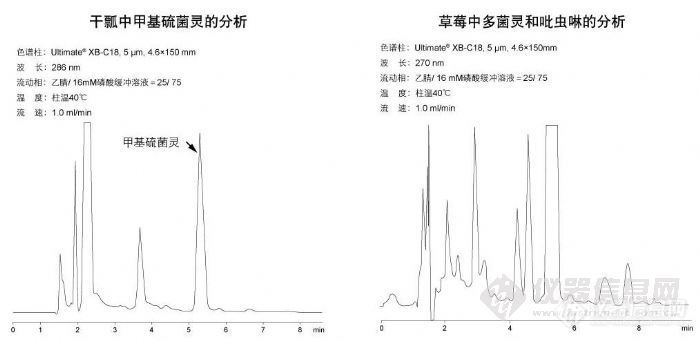
干瓢中甲基硫菌灵的分析与草莓中多菌灵和吡虫啉的分析http://ng1.17img.cn/bbsfiles/images/2009/11/200911021912_180192_1896702_3.jpg
请问 哪位老师做过草甘膦和氨甲基膦酸[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS检测,求前处理和仪器检测方法?
草甘膦和氨甲基膦酸与三氟乙酸酐和七氟丁醇衍生反应,到底生成的衍生物分别是什么?
请教[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]高手关于间甲基苯酚与对甲基苯酚的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析: 我是一个[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]新手,我的目的是用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析间甲基苯酚与对甲基苯酚的混合物,两组分沸点201.8和202.1度,用的是毛细管柱惠普的(BP 20),可是分不开,我用程序升温柱温50~220度,3度每分钟,载气用氮气,柱前压力0.14mpa,进样室250度,检测器250度,分析n次总是出来一个峰,快绝望了,望高手指点,可以给我发邮件chaochaoqunqun@163.com,小弟不胜感激!!!!!!!!
别名:百里香【来源】为唇形科植物麝香草的全草。5~6月采收。【原形态】灌木状常绿草本。茎坚硬直立,四棱形,高18~30厘米,多分枝。叶无柄,对生,线状披针形至卵状披针形,长9~12毫米,宽约4毫米,先端尖,叶缘稍反卷,全缘,基部广楔形,上面具短茸毛,并密生腺点。枝梢疏生轮伞花序;花萼表面有短柔毛及腺点,绿色,下唇2裂成针刺状,上唇3裂,裂片较下唇裂片为短;花冠粉红色,比花萼稍长,上唇直立,油腺明显,有樟脑香味;雄蕊2强,超出花冠,花药红色;雌蕊柱头2裂,红色。小坚果棕褐色。花期5~6月。【生境分布】原产地中海沿岸;我国有栽培。【化学成份】盛花期的全草含挥发油0.8~1.2%。挥发油中含百里香酚(即麝香草脑,为油中的主要成分)、香荆芥酚、对-聚伞花素、l-α-蒎烯、γ-松油烯、α-松油醇,l-脑、石竹烯、芳樟醇、乙酸芳樟醇酯、乙酸龙脑酯、2-甲基-6-亚甲基庚二烯-2,7-醇及其乙酸酯。另有谓不同之品种其挥发油的单萜成分不同,可分别为牻牛儿醇、芳樟醇、α-松油醇、香荆芥酚、百里香酚或侧柏醇-4及松油醇-4。全草尚含:皂甙、熊果酸、齐墩果酸、咖啡酸、绿原酸、喹啉-4-羧酸,少量黄酮类化合物,如木犀草素-7-β-葡萄糖甙、木犀草素-7-二葡萄糖甙等。【药理作用】①抗菌作用挥发油,特别是麝香草脑有防腐、消毒作用,可用于口腔、咽喉之灭菌。麝香草脑作用性质与酚类似,而作用更强(酚系数为25),但遇有机物则效力大减,水中溶解度亦远较酚为差(1/100)。对金黄色葡萄球菌、大肠杆菌均有抑制作用:开花季节茎、叶的乙醇或生理盐水的提取物效力较好。挥发油的蒸汽亦有良好功效。用纸层析可分别获得对革兰氏阳性及革兰氏阴性有效的物质(非鞣质或酚性化合物),在1:300000~500000浓度时即能抑菌。它还有抗真菌作用,1%麝香草脑的醇溶液或2%粉撒布可用于皮肤癣菌病。对放线菌病可以局部应用及口服,但青霉素的疗效较其优越。②驱虫作用麝香草脑对钩虫、鞭虫有麻痹作用,但因其毒性较大,已为其他更好的驱虫剂所取代。尚可用于治疗球虫病。③对呼吸道的作用叶有祛痰作用。其提取物能增进支气管粘膜的分泌,麝香草脑、香荆芥酚能促进纤毛(蛙气管)运动,并以原形自肺排出,故有杀菌作用,有人认为可用于治疗气管炎、百日咳。④其他作用提取物能显著抑制乙酰胆腻引起的豚鼠小肠收缩,而对抗5-羟色胺的作用却较弱,对缓动素之对抗则更差,对组织胺的作用仅有轻微影响。对离体大鼠子宫有兴奋作用,阿托品不能阻止此兴奋。对豚鼠肺灌流时,提取物有轻度松弛支气管的作用,对大鼠血压则无影响。有人称其有驱风作用,内服可治腹泻及鼓肠(减轻肠管发酵),或云可用于通经,并有镇静作用,在体外,它能抑制胆碱酣酶。对家兔静脉注射,可降低血磷,升高血糖。⑤体内过程与毒性麝香草脑在肠胃道可迅速而完全的被吸收;油、酒可促其吸收。半数在体内破坏,其余部分与硫酸根或葡萄糖醛酸相结合而由尿排出。涂于皮肤无刺激性,亦不吸收,但能刺激粘膜,故可引起呕吐,其毒性仅及酚的1/4。口服1克不致引起中毒症状,量大则可致眩晕、上腹剧痛、兴奋、恶心及呕吐、虚弱、流涎流汗、发绀、体温降低、脉细速、呼吸慢乃至昏迷。香荆芥酚与麝香草脑为异构体,作用亦相似,抗菌效力较强,刺激性亦较强;易吸收,可引起呕吐、腹泻,口服致死量在0.1~1克/公斤之间,由于肝脏变性,可于数日或数周内死亡。
本人最近在做职业卫生草酸的方法验证,参考的标准是GBZ/T 300.114-2017,这个标准采用的色谱柱是阳[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱和邻苯二甲酸流动相,我们平时做环境中的草酸都是阴[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱,碳酸钠和碳酸氢钠流动相。大家做职业卫生的草酸的时候具体是怎么操作的?严格按照标准用阳[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]柱做?还是用阴离子做,再写一份方法偏离?先谢谢大家了!
麝香草酚法测硝态氮,加入9毫升氨水后,为什么会出现如图所示的结晶现象呢?请大神给予指导。[img]http://ng1.17img.cn/bbsfiles/images/2017/12/201712072311_01_3345201_3.jpeg[/img]
看一篇关于一种除草剂(灭草松)的农残检测方法,其中前处理说到要用重氮甲烷进行甲基化以后才能进GC—ECD检测,想请教大家的是甲基化的作用是什么?为什么一定要甲基化这个步骤?
用液相原子荧光检测甲基汞时,甲基汞与二价汞分不好,请教大家都用的哪家的柱子什么规格的?流动相比例?还有一个问题是L-半胱氨酸要求是生化试剂,不知道在哪里能买到,用优级醇代替可以吗?请路过的高手指点
含有三氟甲基的化合物其碳谱裂分有什么规律吗?
SNT 2324-2009 进出口食品中抑草磷、毒死蜱、甲基毒死蜱等33种有机磷农药残留量的检测方法
百里香酚兰,百里草酚兰,麝香草酚兰是同一物质吗?请大师给出它们的结构式。谢谢。
最近在做草甘膦,主要是参照SN /T 1923-2007进出口食品中草甘膦残留量的检测方法LCMSMS法,依照标准衍生,标准溶液中加入硼酸和衍生试剂FMOC-CL,上质谱,正离子模式,根本找不到392目离子,衍生试剂的浓度增加了,也没有作用,不知道问题出在哪里了?很是头痛!那位高人做过,请指教!十分感谢!很着急啊做不出来。希望做过的前辈指教。
[color=#444444]2,4-二甲基苯硫酚,2,6-二甲基苯硫酚位置异构体。沸点分别为207-208 °C,76℃[/color][color=#444444]使用Scientific TRACE TR-5MS 色谱柱,两个峰出在一起,相差0.1min,无法基线分离。尝试多种升温程序,只有保留时间和峰宽有变化,分离情况不变。降低载气流速也一样。[/color][color=#444444]换用 安捷伦vf-WAXms柱,彻底分不开了。瘦高的一个峰。[/color][color=#444444]两个柱子都试过60,70,90,120度恒温,分不开。梯度升温,也分不开。[/color][color=#444444]请各位大侠帮帮忙,试了两天了。[/color]
[color=#444444]冰淇淋生产用的奶粉怎样检测抗生素?[/color][color=#444444]抗生素检测[/color][color=#444444]试剂那里有的卖?[/color][color=#444444]加急!![/color][color=#444444]感谢!![/color]
羧甲基淀粉取代度如何测定?除了滴定法,有没有更好的可以快速测定的方法
依据98/34/EC指令的要求,欧盟成员国在将新的技术法规草案和快速社会服务措施写进国家法案前,必须正式告知委员会和其他公众。 3月1日,比利时政府正式发布双酚A禁令法律草案通知,延续1月份比利时参议院投票通过的相关议案:于2013年1月1日起禁止含有双酚A的供3岁以下儿童使用的食品容器(含婴儿奶瓶)的生产制造、置于市场销售。指令向委员会和公众正式通告时间为2012.3.1~2012.6.4。 2011年3月1日,欧盟委员会从当天起禁止含双酚A的婴幼儿奶瓶,从6月1日起成员国禁止进口此类奶瓶。两相对比,比利时的双酚A禁令则将禁止产品类别扩大到3岁以下儿童使用的食品容器,监管范围更大。与之相配合的,双酚A禁令将置于下列指令之下:·(EC) No. 315/93 食品污染物条例;·2000/13/EC食品的标签、展示和宣传指令;·93/43/EEC食品卫生条例;·1935/2004/EC食品接触材料安全条例 通知发出后,即有绿色环保组织呼吁比利时应该将双酚A禁令从2014年起扩大到所有的食品接触材料。资料表明,双酚A属低毒性化合物。动物实验发现双酚A有模拟雌激素的效果,即使很低的剂量也能使动物产生雌性早熟、精子数下降、前列腺增生等作用。此外,双酚A具有一定的胚胎毒性和致畸性,可明显增加动物卵巢癌、前列腺癌、白血病等癌症的发生。双酚A的“江湖封杀令”愈来愈烈,瑞旭技术再一次提醒相关企业:1. 从源头上控制双酚A的使用,加强原料控制,提高自我监督意识;2. 积极研发新技术,寻找替代原料;3. 与国外客户保持良好的沟通,掌握出口目的地法规要求并及时关注国外相关法规动态,提前做好应对。
荠菜古称“护生草”,作为一种野菜,做饺子馅、煮粥、煲汤均可,美味可口,营养丰富。中医认为荠菜具有清肝调脾、和血利水、降压明目的功效。
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定灭草松2.4-滴 标品衍生时需要注意什么 做了衍生 衍生时温度时间都控制了 但还是不出峰
今天配制过程如下: 移取100uL三甲基氯矽烷(TMS)入1.8mL进样小瓶,然后再移取300uL六甲基二矽烷(HMDS)入之,一切正常,但是再移取900uL吡啶入之时,先出现“白雾”,然后再出现固态“沉淀”。 奇怪的是这沉淀漂在上面,振摇,沉淀减少,而且底部的衍生试剂开始澄清。 这正常吗?
按NY/T1096-2006《食品中草甘膦残留量测定》草甘膦衍生时一定要经过干冰浴(-40- -60摄氏度)吗?是否可以绕过此步骤?原理是什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP