苯乙酸是医药、农药、香料等有机合成的中间体。在医药工业中用于青霉素、地巴唑等药物的生产。苯乙酸经氯化、酯化得到α-氯代苯乙酸乙酯,用于稻丰散和乙基稻丰散的生产,这两种农药是广谱性有机磷杀虫剂。苯乙酸本身也是农药植物生长激素。苯乙酸广泛存在于葡萄、草莓、可可、绿茶、蜂蜜等中。苯乙酸在低浓度时具有甜蜂蜜味,在1ppm以下仍具有甜味,是一种重要的香料成分。苯乙酸还具有很强的杀菌作用。
求助各位同行:苯乙酸的国标或行标(HGB3444-62)全文.多谢了.我的邮箱:zjp9933@163.com
[color=#444444]甲酰基苯乙酸甲酯的色谱含量测试中。由于甲酰基苯乙酸甲酯会有醇酮互变性质,在液相色谱(乙腈:水=50:50)下出现三个峰。但是在LC——MS下从第一个峰开始到第三个峰这之间所有的时间都出现了M+1峰(包括峰之间的)。谁能告诉我这是怎么回事。[/color][color=#444444]有哪个大侠做过甲酰基苯乙酸甲酯的含量测试的,求指导。[/color]
目前所做项目需要液相检测联苯乙腈(原料)和联苯乙酸(产物),但两者出峰时间一致,已尝试多种方法进行混合样分离,但始终只有一个尖峰显示,没有任何分离趋势,目前试过的方法有:甲醇:0.05%磷酸水=70:30(C-18长柱);乙腈:水=70:30;乙腈:0.1%氨水水溶液=70:30(NX-C18);甲醇:0.1%醋酸水=55:45(C18短柱)。求助是否有可行的方法能够较好的分离两种物质。谢谢。
如题,在合成中有一步反应是用邻羟基苯乙酸制备其二钠盐,其中产物中可能含有的成分有邻羟基苯乙酸、邻羟基苯乙酸的一钠盐、二钠盐,请问如何建立检测方法将其分离呢?谢谢! 试过液相的方法,但是分不开,也试过双相滴定,但是里面还有过量的NaOH,影响结果,也试过用酚羟基的显色反应,但这个又太灵敏了,无法定量。请大家指导一下吧。
[color=#444444]求助一下啊!!!![/color][color=#444444]我做的羟基苯乙酸去送样,结果GC分析人员做出了两个峰,做了两次都是这样。[/color][color=#444444]我的样品应该是纯的,是分析方法不对,还是其它什么原因啊?有没有哪个遇到这个的情况呢?求助哈[/color]
[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#333333]4-羟基苯乙酸酯[/color][/size][/font],[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]CAS No.:[b]58556-55-1[/b][/color][/size][/font][font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]分子式:C[sub]1[/sub][sub]0[/sub]H[sub]1[/sub][sub]2[/sub]O[sub]3[/sub][/color][/size][/font]
完全按GB29708-2013的方法处理,衍生化后质谱检不出五氯苯乙酸酯,不知道哪里有问题!请做过这个项目的指点。
[color=#d40a00][size=2]维权声明:本文为[font=Times New Roman]11093661[/font]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/size][/color] 青霉素发酵过程中利用液相分析对发酵液中的苯乙酸,效价检测存在着很是矛盾的主体,那就是发酵过程中的效价检测是批量检测,在检测过程中青霉素效价有高有低,这样就不利于药典中规定的标准品与待测品的含量大致相同的规定,这就不可能每做一个样品就做一个标准,这样不实际也不利于节约成本的。那么就需要我们做一个线性范围来使在实际检测的过程中的效价范围在线性范围类,这样就有利于测样的准确性。本人通过长期反复工作实践发现在青霉素发酵过程中发酵液的稀释倍数在100倍以上时,出现了这样一个情况:在检测效价的过程中对苯乙酸检测的结果很不稳定,特别是长期工作的液相更是如此,本人在工作中试验发现原因主要是基线的漂移造成了这个现象。而检测苯乙酸对生产发酵中的地位相当重要,苯乙酸是青霉素发酵过程中的主要原材料之一,而苯乙酸的多少又决定了发酵水平高低。所以说苯乙酸的检测也同样重要。 而效价,苯乙酸是同时检测出来的,如果稀释倍数大于100后解决了效价检测的准确性得到了提高,但是苯乙酸的检测准确性也就低了。所以这个矛盾主体也就出现了。本人在长期的检测中实践发现如果分开来检测,也就是两次检测,而对于两次检测过程中的效价与苯乙酸两种物质稀释倍数采取不同的稀释倍数这样有利于检测结果的准确性。或者对苯乙酸检测采取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测同时稀释倍数根据苯乙酸残量的多少采取不同的稀释倍数这样有利于检测结果的准确性,重现性。而根据本人了解某特大型青霉素发酵公司也就是只是对效价采取的稀释倍数的不同,这样提高了效价的准确性而导致了苯乙酸残量的检测的准确性也就降低了,导致发酵水平无法与成都某特大型青霉素发酵公司的水平相比较。这就说明了苯乙酸检测尤为重于效价检测。而分两次检测或气,液相同时检测这样不利于成本降低,本人通过长期的摸索,比较发现利用苯乙酸的多少来决定稀释倍数后,再根据效价的高低采取不同的标准品的含量,这样就利于检测的准确性与成本的降低——相当于分段检测。 本人事先声明这个纯属个人自己摸索试验得出的结论,在实际生产,检测中虽然得到了应用,但由于自己的试验结果,水平有限所以具体过程没有完全介绍,十分抱歉。
[color=#444444]乙酸对硝基苯酯与甲醇反应,监测反应进程的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分离条件该怎么设?[/color][color=#444444]查文献查不到具体的色谱条件,进样口温度,柱温,检测器温度以及对[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱有无要求?[/color][color=#444444]已知:乙酸对硝基苯酯沸点为297℃;对硝基苯酚沸点为279℃;乙酸甲酯沸点为57.8℃;甲醇沸点为64.7℃。[/color]
间硝基苯乙酮、CAS:121-89-1 质量标准 化学试剂 工业级均可,请大家帮帮忙。
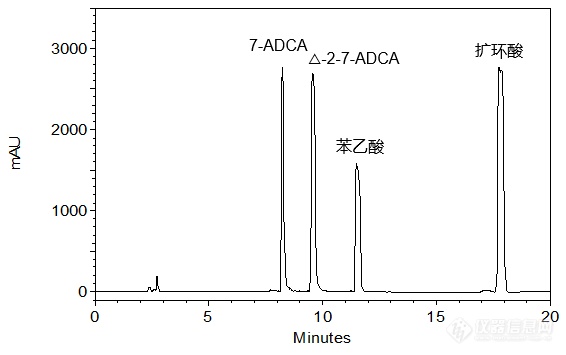
[align=center][b]4种头孢中间体的共同分析[/b][/align][align=right][b]——7-ADCA、△-2-7-ADCA、苯乙酸及扩环酸的分析[/b][/align][align=right][b][/b][/align]客户提供了7-ADCA(7-氨基去乙酰氧基头孢烷酸),△-2-7-ADCA、苯乙酸及扩环酸原料,希望本实验室依据客户所提供的色谱条件筛选合适的C[sub]18[/sub]色谱柱,实现以上4种化合物的稳定良好分析。本实验室参考客户提供的色谱条件,首先尝试使用经过聚合物包被处理的中等极性色谱柱——CAPCELL PAK C[sub]18[/sub] MGII对客户所提供样品进行分析,使用PDA检测器进行检测。如图1,在高浓度大体积进样的情况下,各色谱峰发生严重过载现象,出现平头峰;如图2,降低进样体积至5 μL可得到相对良好峰形,且各组分间能够得到良好分离,分离度均在10.0以上(结果详见表1)。[align=center][img=,566,355]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537444868_8925_2222981_3.png!w566x355.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,575,365]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545063985_5910_2222981_3.png!w575x365.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表1 CAPCELL PAK C[sub]18[/sub] MGII分析结果详表(进样量:5 μL)[/align][align=center][img=,553,136]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537465105_7074_2222981_3.png!w553x136.jpg[/img][/align][align=center][/align][align=left][img=,579,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545307700_4011_2222981_3.png!w579x319.jpg[/img][/align][align=left][/align][align=left]为使客户有更多色谱柱选择,本实验室也尝试了能够在纯水系流动相下稳定使用的高极性色谱柱——CAPCELL PAK C[sub]18[/sub] AQ进行分析。如图3,几种头孢中间体的整体保留有所增强,而高浓度上样仍会出现与MGII色谱柱相似的过载现象;如图4,降低进样体积进行分析,可得到良好结果,同时发现扩环酸有一定程度的拖尾(见表2)。[/align][align=center][/align][align=center][img=,527,377]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546103944_1716_2222981_3.png!w527x377.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,525,374]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546123571_3070_2222981_3.png!w525x374.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表2 CAPCELL PAK C[sub]18 [/sub]AQ分析结果详表(进样量:5 μL)[/align][align=center][img=,558,135]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546125551_7090_2222981_3.png!w558x135.jpg[/img][/align][align=center][/align][align=left][img=,577,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101547424196_8227_2222981_3.png!w577x319.jpg[/img][/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 250 mm和高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 250 mm,以磷酸盐缓冲液(pH 6.0)-乙腈为流动相体系,在30°C柱温条件下进行梯度分析,均能够实现7-ADCA、△-2-7-ADCA、苯乙酸和扩环酸的良好分离,其中,CAPCELL PAK C[sub]18[/sub] MGII色谱柱所得峰形更佳。[/align]
我想用毛细管GC测苯乙烯、二乙烯苯,何如选择固定相及其他实验条件。要测硝基苯中甲酰胺、甲酸等。
参考GB/T 2912.3-2009 纺织品甲醛的测定 高效液相色谱法其中衍生化试液配制方法如下:称取0.05g 2,4-二硝基苯肼,用适量内含0.5%乙酸的乙腈溶解后置于100ml容量瓶中,用水稀释至刻度。据我了解,目前市场上提供2,4-二硝基苯肼,加有30%或者50%的水作为稳定剂,所以产品看上像是泥水混合物,有粘稠状的固体,也有流动的水,对于上述配置过程中称量50mg的2,4-二硝基苯肼,该如何称量?是称50mg还是100mg就可以了?有么有做过实验的说一下经验哦。我看过也有用25%乙腈水溶液对2,4-二硝基苯肼重蒸提取的,然后再称量。
当前开展丙二醛 2,4-二硝基苯肼柱前衍生液相色谱310nm检测,流动相0.2%乙酸-乙腈(1:1),结果只有 2,4-二硝基苯肼峰,未见明显衍生产物峰,没有衍生好是什么原因。衍生方法为丙二醛在高氯酸或三氯乙酸酸性条件衍生。
请问各位,硝基苯的标准溶液配置中,称出硝基苯的质量,怎么算浓度。最后算出硝基苯的吸光度和苯胺的吸光度,两者相减对应的含量即为m,这个对应的含量从哪里查到还是自己算?有没有公式?C=m*5/V不会算呐
[table=100%][tr][td]想用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]测水中的水中苯酚,苯,硝基苯,但在用二氯甲烷将三种有机物的混标稀释到1ppm,0.1ppm,0.01ppm,0.001ppm几个不同浓度梯度的混合使用液时,[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测却不出峰,用乙酸乙酯稀释标液测试也不出峰,不知道哪出问题了,机子也没问题啊,求大神解答?[/td][/tr][/table]

[color=#DC143C]硝基苯[/color][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911192347_185515_1610969_3.jpg[/img]硝基苯 又名密斑油,苦杏仁油 CAS:98-95-3 英文名:nitrobenzene 无色或微黄色具苦杏仁味的油状液体。(纯净应为无色,实验室制硝基苯由于溶有硝酸分解产生的二氧化氮而有颜色,除杂方式:加氢氧化钠溶液 分液) 分子式C6H5NO2(结构如图)。分子量123.11。相对密度1.205(15/4℃)。熔点5.7℃。沸点210.9℃。闪点87.78℃。自燃点482.22℃。蒸气密度4.25。蒸气压0.13kPa(1mmHg44.4℃)。难溶于水,密度比水大 易溶于乙醇、乙醚、苯和油。遇明火、高热会燃烧、爆炸。与硝酸反应剧烈。 实验室制法:目的原理 主反应: Ar + HONO2 +H2SO4 Ar- NO2 + H2O 副反应: Ar- NO2+ HONO2 +H2SO4 Ar-(NO2)2+ H2O 仪器药品 苯8.9ml (7.8g,0.1mol),硝酸(d = 1.40) 7.3ml (0.11mol),浓硫酸(d = 1.84) 10ml (0.18mol),10%碳酸钠溶液,饱和食盐水,无水氯化钙。 过程步骤 在50ml圆底烧瓶上装配一个二口连接管,正口配一温度计,其水银球离瓶底约5mm,侧口装配一回流冷凝管。也可以用一个二口烧瓶,正口装配回流冷凝管,侧口装一温度计,其水银球离瓶底约5mm。在烧瓶中加入8.9ml苯。通过冷凝管上口,将已冷却的混酸分多次加入苯中。每加一次后,必须充分振荡烧瓶,使苯与混酸充分接触,待反应物的温度不再上升而趋于下降时,才继续加混酸(为什么?)。反应物的温度应保持在40~50℃之间,若超过50℃,可用冷水浴冷却烧瓶。加料完毕后,把烧瓶放在水浴上加热,约于10min内把水浴加热到60℃(反应混合物的温度为60~65℃)并保持30min,间歇地振荡烧瓶。 冷却后,将反应混合物倒入分液漏斗中。静置分层,分出酸层(哪一层?怎样判断和检验?),倒入指定回收瓶内。粗硝基苯先用等体积的冷水洗涤,再用10%碳酸钠溶液洗涤,直到洗涤液不显酸性。最后用水洗至中性(如何检验?)。分离出粗硝基苯,放在干燥的小锥形瓶中,加入无水氯化钙干燥,间歇振荡锥形瓶。 把澄清透明的硝基苯倒入30ml蒸馏烧瓶中,连接空气冷凝管。在石棉网上加热蒸馏,收集204~210℃的馏分。为了避免残留在烧瓶中的二硝基苯在高温下分解而引起爆炸,注意切勿将产物蒸干。 产量:约9.5g。 纯硝基苯为无色液体,具有苦杏仁气味,沸点210.9℃,d20= 41.203。
参考GB/T 2912.3-2009 纺织品甲醛的测定 高效液相色谱法其中衍生化试液配制方法如下:称取0.05g 2,4-二硝基苯肼,用适量内含0.5%乙酸的乙腈溶解后置于100ml容量瓶中,用水稀释至刻度。据我了解,目前市场上提供2,4-二硝基苯肼,加有30%或者50%的水作为稳定剂,所以产品看上像是泥水混合物,有粘稠状的固体,也有流动的水,对于上述配置过程中称量50mg的2,4-二硝基苯肼,该如何称量?是称50mg还是100mg就可以了?有么有做过实验的说一下经验哦。我看过也有用25%乙腈水溶液对2,4-二硝基苯肼重蒸提取的,然后再称量。
硝基苯的制备目的原理主反应: Ar + HONO2 +H2SO4 Ar- NO2 + H2O副反应: Ar- NO2+ HONO2 +H2SO4 Ar-(NO2)2+ H2O仪器药品苯8.9ml (7.8g,0.1mol),硝酸(d = 1.40) 7.3ml (0.11mol),浓硫酸(d = 1.84) 10ml (0.18mol),10%碳酸钠溶液,饱和食盐水,无水氯化钙。过程步骤在50ml圆底烧瓶上装配一个二口连接管,正口配一温度计,其水银球离瓶底约5mm,侧口装配一回流冷凝管。也可以用一个二口烧瓶,正口装配回流冷凝管,侧口装一温度计,其水银球离瓶底约5mm。在烧瓶中加入8.9ml苯。通过冷凝管上口,将已冷却的混酸分多次加入苯中。每加一次后,必须充分振荡烧瓶,使苯与混酸充分接触,待反应物的温度不再上升而趋于下降时,才继续加混酸(为什么?)。反应物的温度应保持在40~50℃之间,若超过50℃,可用冷水浴冷却烧瓶。加料完毕后,把烧瓶放在水浴上加热,约于10min内把水浴加热到60℃(反应混合物的温度为60~65℃)并保持30min,间歇地振荡烧瓶。冷却后,将反应混合物倒入分液漏斗中。静置分层,分出酸层(哪一层?怎样判断和检验?),倒入指定回收瓶内。粗硝基苯先用等体积的冷水洗涤,再用10%碳酸钠溶液洗涤,直到洗涤液不显酸性。最后用水洗至中性(如何检验?)。分离出粗硝基苯,放在干燥的小锥形瓶中,加入无水氯化钙干燥,间歇振荡锥形瓶。把澄清透明的硝基苯倒入30ml蒸馏烧瓶中,连接空气冷凝管。在石棉网上加热蒸馏,收集204~210℃的馏分。为了避免残留在烧瓶中的二硝基苯在高温下分解而引起爆炸,注意切勿将产物蒸干。产量:约9.5g。纯硝基苯为无色液体,具有苦杏仁气味,沸点210.9℃,d20= 41.203。注意事项1.苯的硝化反应也可在三口烧瓶中进行。在100ml三口烧瓶中放入苯,在中间瓶口安装搅拌棒,一个侧口装上冷凝管,另一侧口插上温度计,其水银球要浸到液面下。开动搅拌器,从冷凝管上口分批加入已冷却的混酸。其余的步骤与用圆底烧瓶时一样。全部药品用量都加倍。2.混酸配制法:在50ml锥形瓶中放入10ml浓硫酸,把锥形瓶置于冷水浴中,一边不停地摇动锥形瓶,一边将7.3ml硝酸慢慢地注入浓硫酸中。3.苯的硝化反应为一放热反应。在开始加入混酸时,硝化反应速率较小,每次加入的混酸量宜为0.5~1ml。随着混酸的加入和硝基苯的生成,反应混合物中的苯的浓度逐渐降低,硝化反应的速率也随之减小,故在加入后一半混酸时,每次可加入1.5~2ml。4.用吸管吸取少许上层反应液,滴到饱和食盐水中,当观察到油珠下沉时,那就表示硝化反应已经完成。5.硝基苯有毒,处理时须加小心。如果溅在皮肤上,可先用少量酒精洗擦,再用肥皂水洗净。6.如果使用工业浓硫酸,其中含有的少量汞盐等杂质具有催化作用,使反应产物中含有微量的多硝基酚,如苦味酸和2,4-二硝基苯酚,它们的碱溶液呈深黄色。应洗到碱溶液几近无色。分析思考 1.硫酸在本实验中起什么作用?2 .一次把混酸加完,会产生什么结果?3.若用相对密度为1.52的硝酸来配制混酸进行苯的硝化,将得到何产物?
水质硝基苯的测定锌还原分光光度法中,加入硫酸氢钾后沉淀不消失,这是什么原因导致的呢?
我们做水中的二硝基苯,有三种同分异构体,邻,间,对-二硝基苯,都有不同的检出限,但我们的报告上只需要报二硝基苯的总量,那检出限应该怎么写呢?是写其中最小的,或最大的,还是三个同分异构体的总和?
事件回放:“丈夫没有了,两个孩子也还在医院里,这两天我都不知道怎么过来的。”刘从兰用手擦去眼角的眼泪,静静地望着刚做完血透的小女儿琳琳(化名)。她说,现在一点都不敢想象今后的生活该怎么过下去,怕自己会承受不了。 9月13日,东阳市画水镇发生一起因废塑中毒事件,目前已造成3人死亡、17人住院治疗的严重后果。[font=黑体][size=4]何谓二硝基苯酚?[/size][/font][color=#00008B]分子式2,4-(NO2)2C6H3F。 2,4-二硝基氟苯为淡黄色晶体; 熔点25.8℃,沸点 296℃,密度1.4718克/厘米3(84℃); 溶于乙醇、苯、丙二醇等。 2,4-二硝基氟苯主要由2,4-二硝基氯苯与氟化钾在硝基苯中反应制得 2,4-二硝基氟苯是一种重要的分析试剂,用来鉴定有机化合物中的氨基,尤其是用于蛋白质或多肽的N-端残基分析。鉴定时,2,4-二硝基氟苯与肽链的游离氨基作用,生成2,4-二硝基衍生物。将其水解后,末端氨基酸的N-(2,4-二硝基苯基)衍生物常为亮黄色结晶,易与其他氨基酸分离。该方法结合其他方法,可确定蛋白质或多肽氨基端碳链的结构。由F.桑格于1945年提出,故称桑格法 此外,它在碳酸氢钠溶液中与醛糖的肟反应时可发生降解,生成次级醛糖、2,4-二硝基苯酚和氢氰酸,故可用于醛糖的分析。2,4-二硝基氟苯能使皮肤糜烂,使用时应注意。[/color]&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&77[color=#DC143C]1)作为一名专业的分析工作者,如果从自己的专业角度去分析。我们使用什么方法和仪器可以检测出二硝基苯酚?2)遇到这样的事情如何加强自我预防?3)在我们平时的分析工作中,我们会遇到那些有毒的化学试剂或者样品,我们如何做好自我防备。欢迎大家讨论。。。。[/color]
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能够将间位硝基苯酚、对位硝基苯酚、邻位硝基苯酚能分开么?谢谢
求助:乙醛与2,4-二硝基苯肼为什么反应不完全。用的稀释剂是乙腈:水(1:1)。如果有的话请提供详细衍生方法,如水浴温度,衍生时间及酸的浓度。谢谢。
大家好,请问对二硝基苯和邻二硝基苯的纯物质可以溶于甲醇吗?请问有谁试过,主要是想配混标,其中间二硝基苯买的标液是甲醇中的标液。
请教大家:有没有使用过HJ 716-2014方法测定水中硝基苯类化合物的(主要项目为:硝基苯,间、对、邻硝基氯苯,间、对、邻二硝基苯,2,4-二硝基氯苯,2,4-二硝基甲苯,2,4,6-三硝基甲苯)?有几个问题请教一下,第一:我使用的是HP-5MS的色谱柱,但是硝基苯和内标硝基苯D5分不开,硝基苯的线性做的很不好,硝基甲苯的同分异构体线性都不是很好;第二:低浓度0.1mg/L是不是太低了,有很多的成分都扫不出来,按照标准方法,我用的是SCAN模式,大家都采用什么浓度?第三:用什么方法和仪器测定水中硝基苯类比较好?请各位大神指导
水质 一硝基苯和二硝基苯化合物 还原-偶氮光度法中 加入10%NaOH、20%KHSO4、5%亚硝酸钠、2.5%氨基磺酸按、N-(1-萘基)乙二胺的作用分别是什么?谢谢!!有做过的大神告知一下
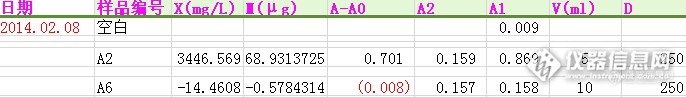
测水中的硝基苯,用的方法是水和废水监测书上的 盐酸萘乙二胺分光法。 水样放15天前后,数据比对,看下图http://ng1.17img.cn/bbsfiles/images/2014/02/201402111525_489908_2721409_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/02/201402111525_489909_2721409_3.png下面我从右到左解释一下:D——代表的是稀释倍数;V(ml)—取样的体积;A1—是还原后硝基苯跟水样原有苯胺的总吸光度值;A2—是水样原有苯胺的吸光度值;M—曲线值;X(mg/L)—水样中含有硝基苯的量。前后数值诡异的让我难以置信。各专家们围观一下,看会不会有这样的情况?我是哪里没做好吗
请教各位大虾:有没有成型的液相色谱测定硝基苯的方法,紫外检测器。方法回收率,标准偏差经过验证的,可否共享一下。还想请问,有现成的水及鱼类体内残留硝基苯前处理和富集的方法没?







 我要推广仪器
我要推广仪器
 下载APP
下载APP