苯乙酸是医药、农药、香料等有机合成的中间体。在医药工业中用于青霉素、地巴唑等药物的生产。苯乙酸经氯化、酯化得到α-氯代苯乙酸乙酯,用于稻丰散和乙基稻丰散的生产,这两种农药是广谱性有机磷杀虫剂。苯乙酸本身也是农药植物生长激素。苯乙酸广泛存在于葡萄、草莓、可可、绿茶、蜂蜜等中。苯乙酸在低浓度时具有甜蜂蜜味,在1ppm以下仍具有甜味,是一种重要的香料成分。苯乙酸还具有很强的杀菌作用。
求助各位同行:苯乙酸的国标或行标(HGB3444-62)全文.多谢了.我的邮箱:zjp9933@163.com
[color=#444444]甲酰基苯乙酸甲酯的色谱含量测试中。由于甲酰基苯乙酸甲酯会有醇酮互变性质,在液相色谱(乙腈:水=50:50)下出现三个峰。但是在LC——MS下从第一个峰开始到第三个峰这之间所有的时间都出现了M+1峰(包括峰之间的)。谁能告诉我这是怎么回事。[/color][color=#444444]有哪个大侠做过甲酰基苯乙酸甲酯的含量测试的,求指导。[/color]
目前所做项目需要液相检测联苯乙腈(原料)和联苯乙酸(产物),但两者出峰时间一致,已尝试多种方法进行混合样分离,但始终只有一个尖峰显示,没有任何分离趋势,目前试过的方法有:甲醇:0.05%磷酸水=70:30(C-18长柱);乙腈:水=70:30;乙腈:0.1%氨水水溶液=70:30(NX-C18);甲醇:0.1%醋酸水=55:45(C18短柱)。求助是否有可行的方法能够较好的分离两种物质。谢谢。
如题,在合成中有一步反应是用邻羟基苯乙酸制备其二钠盐,其中产物中可能含有的成分有邻羟基苯乙酸、邻羟基苯乙酸的一钠盐、二钠盐,请问如何建立检测方法将其分离呢?谢谢! 试过液相的方法,但是分不开,也试过双相滴定,但是里面还有过量的NaOH,影响结果,也试过用酚羟基的显色反应,但这个又太灵敏了,无法定量。请大家指导一下吧。
[color=#444444]求助一下啊!!!![/color][color=#444444]我做的羟基苯乙酸去送样,结果GC分析人员做出了两个峰,做了两次都是这样。[/color][color=#444444]我的样品应该是纯的,是分析方法不对,还是其它什么原因啊?有没有哪个遇到这个的情况呢?求助哈[/color]
[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#333333]4-羟基苯乙酸酯[/color][/size][/font],[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]CAS No.:[b]58556-55-1[/b][/color][/size][/font][font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]分子式:C[sub]1[/sub][sub]0[/sub]H[sub]1[/sub][sub]2[/sub]O[sub]3[/sub][/color][/size][/font]
完全按GB29708-2013的方法处理,衍生化后质谱检不出五氯苯乙酸酯,不知道哪里有问题!请做过这个项目的指点。
[color=#d40a00][size=2]维权声明:本文为[font=Times New Roman]11093661[/font]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/size][/color] 青霉素发酵过程中利用液相分析对发酵液中的苯乙酸,效价检测存在着很是矛盾的主体,那就是发酵过程中的效价检测是批量检测,在检测过程中青霉素效价有高有低,这样就不利于药典中规定的标准品与待测品的含量大致相同的规定,这就不可能每做一个样品就做一个标准,这样不实际也不利于节约成本的。那么就需要我们做一个线性范围来使在实际检测的过程中的效价范围在线性范围类,这样就有利于测样的准确性。本人通过长期反复工作实践发现在青霉素发酵过程中发酵液的稀释倍数在100倍以上时,出现了这样一个情况:在检测效价的过程中对苯乙酸检测的结果很不稳定,特别是长期工作的液相更是如此,本人在工作中试验发现原因主要是基线的漂移造成了这个现象。而检测苯乙酸对生产发酵中的地位相当重要,苯乙酸是青霉素发酵过程中的主要原材料之一,而苯乙酸的多少又决定了发酵水平高低。所以说苯乙酸的检测也同样重要。 而效价,苯乙酸是同时检测出来的,如果稀释倍数大于100后解决了效价检测的准确性得到了提高,但是苯乙酸的检测准确性也就低了。所以这个矛盾主体也就出现了。本人在长期的检测中实践发现如果分开来检测,也就是两次检测,而对于两次检测过程中的效价与苯乙酸两种物质稀释倍数采取不同的稀释倍数这样有利于检测结果的准确性。或者对苯乙酸检测采取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测同时稀释倍数根据苯乙酸残量的多少采取不同的稀释倍数这样有利于检测结果的准确性,重现性。而根据本人了解某特大型青霉素发酵公司也就是只是对效价采取的稀释倍数的不同,这样提高了效价的准确性而导致了苯乙酸残量的检测的准确性也就降低了,导致发酵水平无法与成都某特大型青霉素发酵公司的水平相比较。这就说明了苯乙酸检测尤为重于效价检测。而分两次检测或气,液相同时检测这样不利于成本降低,本人通过长期的摸索,比较发现利用苯乙酸的多少来决定稀释倍数后,再根据效价的高低采取不同的标准品的含量,这样就利于检测的准确性与成本的降低——相当于分段检测。 本人事先声明这个纯属个人自己摸索试验得出的结论,在实际生产,检测中虽然得到了应用,但由于自己的试验结果,水平有限所以具体过程没有完全介绍,十分抱歉。
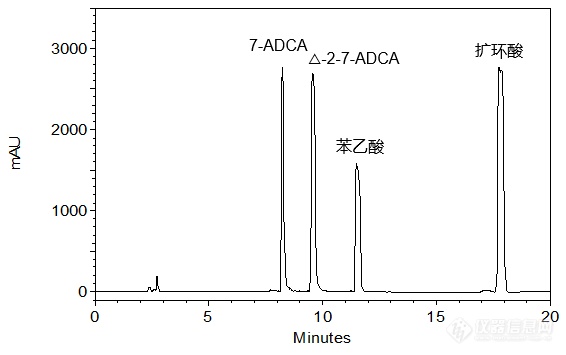
[align=center][b]4种头孢中间体的共同分析[/b][/align][align=right][b]——7-ADCA、△-2-7-ADCA、苯乙酸及扩环酸的分析[/b][/align][align=right][b][/b][/align]客户提供了7-ADCA(7-氨基去乙酰氧基头孢烷酸),△-2-7-ADCA、苯乙酸及扩环酸原料,希望本实验室依据客户所提供的色谱条件筛选合适的C[sub]18[/sub]色谱柱,实现以上4种化合物的稳定良好分析。本实验室参考客户提供的色谱条件,首先尝试使用经过聚合物包被处理的中等极性色谱柱——CAPCELL PAK C[sub]18[/sub] MGII对客户所提供样品进行分析,使用PDA检测器进行检测。如图1,在高浓度大体积进样的情况下,各色谱峰发生严重过载现象,出现平头峰;如图2,降低进样体积至5 μL可得到相对良好峰形,且各组分间能够得到良好分离,分离度均在10.0以上(结果详见表1)。[align=center][img=,566,355]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537444868_8925_2222981_3.png!w566x355.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,575,365]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545063985_5910_2222981_3.png!w575x365.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18 [/sub]MGII色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表1 CAPCELL PAK C[sub]18[/sub] MGII分析结果详表(进样量:5 μL)[/align][align=center][img=,553,136]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101537465105_7074_2222981_3.png!w553x136.jpg[/img][/align][align=center][/align][align=left][img=,579,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101545307700_4011_2222981_3.png!w579x319.jpg[/img][/align][align=left][/align][align=left]为使客户有更多色谱柱选择,本实验室也尝试了能够在纯水系流动相下稳定使用的高极性色谱柱——CAPCELL PAK C[sub]18[/sub] AQ进行分析。如图3,几种头孢中间体的整体保留有所增强,而高浓度上样仍会出现与MGII色谱柱相似的过载现象;如图4,降低进样体积进行分析,可得到良好结果,同时发现扩环酸有一定程度的拖尾(见表2)。[/align][align=center][/align][align=center][img=,527,377]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546103944_1716_2222981_3.png!w527x377.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:50 μL)[/align][align=center][img=,525,374]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546123571_3070_2222981_3.png!w525x374.jpg[/img][/align][align=center]图4 CAPCELL PAK C[sub]18 [/sub]AQ色谱柱分析结果(进样量:5 μL)[/align][align=center] [/align][align=center]表2 CAPCELL PAK C[sub]18 [/sub]AQ分析结果详表(进样量:5 μL)[/align][align=center][img=,558,135]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101546125551_7090_2222981_3.png!w558x135.jpg[/img][/align][align=center][/align][align=left][img=,577,319]http://ng1.17img.cn/bbsfiles/images/2018/05/201805101547424196_8227_2222981_3.png!w577x319.jpg[/img][/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 250 mm和高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 250 mm,以磷酸盐缓冲液(pH 6.0)-乙腈为流动相体系,在30°C柱温条件下进行梯度分析,均能够实现7-ADCA、△-2-7-ADCA、苯乙酸和扩环酸的良好分离,其中,CAPCELL PAK C[sub]18[/sub] MGII色谱柱所得峰形更佳。[/align]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=127755]对羟甲基苯氧乙酸的合成[/url]
请教α-甲基苯乙烯,邻甲基苯乙烯,2-甲基苯乙烯,3-甲基苯乙烯,4-甲基苯乙烯,是不同的物质吗?,物质结构
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
做的五氯苯酚,酯化后上机2ppm 浓度峰强度仅有70000多,远低于基线600000,这是哪里出的问题,方法检出限可是0.05ppm,这是用SIM做的原因吗?
客户要我们产品水杯做LFGB测试项目,我们产品材料是SMM 聚苯乙烯-甲基丙烯酸甲酯,哪位专家老师知道呀?告诉一下。谢谢!
二氯苯甲酸、乙酸等用什么色谱柱?样品中含二氯苯甲酸、乙酸、乙酸丁酯、醋酸铜、四甲基乙二胺、4-甲基吡啶应该怎么处理?用什么色谱柱?用安捷伦6890 FID色谱仪
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
异氰基乙酸乙酯和N,N-二甲基苯胺是否可以用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?有没有方法条件呢?
用YC/T207-2014方法,测混合标液,苯乙烯 2-乙氧基乙基乙酸酯 邻-二甲苯分离不好还,出现了重合峰情况,用谱库搜出来是间二甲苯,邻二甲苯甲苯去哪里了呢?[img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221658210017_6074_5898744_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221658214942_5520_5898744_3.png[/img]
用YC/T207-2014方法,测混合标液,苯乙烯 2-乙氧基乙基乙酸酯 邻-二甲苯分离不好还,出现了重合峰情况,用谱库搜出来是间二甲苯,邻二甲苯甲苯去哪里了呢?![img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221603511087_6024_5898744_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212221603511511_1108_5898744_3.png[/img]
有没有老师做过苯乙醇和乙酸苯乙醇的TLG分离的。在展开剂方面给点意见吧。
本人下午 用亚硝酸纳滴定,用永停滴定法 但是重氮反应较慢。不能明显的判断终点。有什么方法能明显的判断终点吗? 还有就是 中和法,用NAOH滴定能用吗? 滴定终点和化学终点偏差大吗?有哪位知道的 解答下
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
醇类,醚类,酯类,芳烃等16种有机挥发物,2到8个碳已经有了效果很好的分离条件,但是定量方法始终确定不了 (之前没有做过这么多的成分,不知道用内标还是外标合适)希望大家能给个意见,并且希望能说明下选择的原因,谢谢 !!!分离效果图和色谱条件会尽快补上待分析物质如下:乙醇丙酮异丙醇丁酮乙酸乙酯正丁醇苯丙二醇甲醚乙酸正丙酯4-甲基-2-戊酮甲苯乙酸正丁酯乙苯二甲苯环己酮乙酸异丙酯
[color=#444444]我用辛酸甲酯methyloctanoate (C9H18O2) 做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的内标物,绘制不同分析物的标准曲线,各个分析物的相对校正因子差别很大。实验室人告诉我,如果分析物和内标结构差不多,那么校正因子越接近1。这是对的,不过有的化合物化学式差不多,结构却相差很多,这样校正因子差别也很大,我要如何判断我做出来的标准曲线和相对校正因子是对的呢?[/color][color=#444444]比如,我用辛酸甲酯做内标,测了两个化合物,苯乙酸(C8H8O2,含苯环和羧酸)和香兰素(C8H8O3,含苯环,羰基,甲氧基和羟基)。其中苯乙酸相对辛酸甲酯差别不是很大,而香兰素差别就大了。所以他们的标准曲线分别是y=0.7311x-0.0525 R2=0.99998,y-1.1526x-0.1764 R2=0.9982。不知道它们的校正因子对不对?有大神帮忙分析一下吗?[/color]
我用的是据二甲基硅氧烷的柱子,30m*0.25mm*0.25um,溶剂是乙酸乙酯,溶质是苯,两者出峰时间太接近,分离效果很不好进样口:200度检测器:240度程序升温:35度恒温15min各位大大有什么好的方法呢?
请问乙酸的甲基在氘代氯仿中的化学位移在多少啊?谢谢
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
======在 2006-11-13 13:46:31 wangjiangli0123来信中写道:======请问 电离电位是多少 苯乙烯 的 乙酸丁酯的 还有十一烷的======================================
请问乙酸的甲基在氘代氯仿中的化学位移在多少啊?谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP