我想和大家讨论一个关于草甘瞵和草甘瞵铵盐的问题,我公司用草甘瞵合成草甘瞵铵盐,我用液相色谱分析草甘瞵铵盐,但是发现样品溶解性不好,我觉得可能是合成不完全,还有一部分草甘瞵在里面,因为色谱分析只有草甘瞵出峰,铵盐使用分析出来的草甘瞵的含量乘以1.1005后得出的结果,所以导致乘以系数后含量到了100%以上,是不是草甘瞵如果反应彻底的话,应该很好溶解才对,也就是草甘瞵铵盐很好溶解,如果有不溶物,说明肯定有草甘瞵存在,对不对?还有像这种情况用什么方法才能将里面的草甘瞵和草甘瞵铵盐的含量分别检测出来呢?用液谱好象不行吧,因为液谱铵盐是不出峰的,有没有化学方法可以检测出来,也就是说看一看合成效果怎么样?
请问草甘膦和草甘膦铵盐怎样分别检测出含量呢?是混合物
那位大哥大姐能提供一下有关水果中草甘膦铵盐的检测方法吗?还想知道草甘膦的检测方法是不是也可以用来检测草甘膦铵盐?
顶空毛细管[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定甘草酸单铵盐中乙醇残留量,用的是安捷伦7820A,现在有HP-5 DB-1301 DB-1三个柱子,请教一下应该用哪个柱子做乙醇残留啊
谁做过4-磺酸邻苯二甲酸三铵盐含量分析,如何分析?具体条件?
草甘膦、氨甲基膦酸和草甘膦内标衍生: 取1ml 20g/L上述物质标准溶液(水作溶剂),分别加0.5ml 50g/L硼酸钠、0.5ml 10g/LFMOC-Cl衍生剂,37℃衍生2h。SN/T 1923是缓冲盐和衍生剂各加0.2ml,并且衍生剂浓度是1g/L,担心衍生不完全我多加了,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS上扫描啥也没有或者说响应极低不稳定,倒是有几个离子响应很稳,比如草甘膦扫出来是179,365和397,正常来说179、392。 后面,我又直接拿草甘膦和氨甲基膦酸(未经衍生)上机扫描,文献上有负离子扫描出峰的,我试了一下,草甘膦(分子量169)扫出来是168,响应较低不稳定,但合理。就是说仪器没问题,标准品没问题。 问题来了,到底是哪里出了问题?衍生剂我都是临配现用的,顶多也是过量了而已
“农药废渣盐”冒充食盐 跨省域形成私盐贩销链 去年,安徽阜阳盐业和公安部门在公路上截获18吨私盐,并顺藤摸瓜,发现了一起性质恶劣的“农药废渣盐”流入食用市场案件。经查,江苏镇江海天盐化公司非法制售“农药废渣盐”14000吨,流入全国12个省、市盐业市场。该公司并无盐品生产经营资质,其生产原料来自镇江江南化工有限公司生产农药后产生的废渣。 镇江江南化工有限公司主要生产除草剂“草甘磷”,会产生一种污染性很强的工业废渣难以处理。2009年,商人徐敬东、陶先楚、刘伟等人在镇江成立了“海天盐化”公司,从该公司以每吨10元的价格收购农药残渣。 据专案组成员、阜阳市公安局颍泉分局干警王智勇介绍,“海天盐化”在未取得生产、销售工业盐资格的情况下,通过将农药残渣清洗、烘干生产出工业盐。自2009年9月至2011年3月,“海天盐化”共非法制售“农药废渣盐”14000吨。据调查,安徽阜阳截获的“农药废渣盐”,主要卖给当地小商贩用于制作烧饼、馓子等食品。 经中国农业大学分析与环境毒理实验室检验,“农药废渣盐”中的农药“草甘膦”含量高达55毫克/公斤,类似于奶制品中的“三聚氰胺”,明显高于美国、欧盟、日本农产品贸易每公斤20毫克的安全标准。安徽阜阳目前未发现因食用“农药废渣盐”而产生的病例,但有专家认为可能会有潜在性危害。 据调查,“海天盐化”以10元每吨购得原料,生产总成本约每吨100元,以每吨350元至400元的价格卖给私盐批发商;私盐批发商再以每吨700元批发给不法粮油店,粮油店最后以每吨1400元的价格卖给小商贩加工食品。由此,“农药废渣盐”产业链上每个环节都产生了100%或更高的利润率。草甘膦(glyphosate),是由美国孟山都公司开发的除草剂。又称:镇草宁、农达(Roundup)、草干膦、膦甘酸。纯品为非挥发性白色固体,比重为0.5,大约在230℃左右熔化,并伴随分解。25℃时在水中的溶解度为1.2%,不溶于一般有机溶剂,其异丙胺盐完全溶解于水。不可燃、不爆炸,常温贮存稳定。对中炭钢、镀锌铁皮(马口铁)有腐蚀作用。
草甘膦和氨甲基膦酸与三氟乙酸酐和七氟丁醇衍生反应,到底生成的衍生物分别是什么?
最近在做草甘膦。根据SN/T 1923-2007进行衍生。首先衍生了一个5ppm的标液,用于仪器优化,选择合适的锥孔电压和碰撞能,但是衍生完将标液打进质谱时发现。内标分子量应该是395,外标应该是392,但是我的标液打进去,内标外标母离子都在393出很大的峰,395和392没有峰,用的仪器是waters XE有没有知道原因的老师,帮忙解答一下,谢谢
我们一直用气质检测茶叶中草甘膦。但由于要做衍化,前处理比较复杂。为此还专门购进能制冷零下60度的超低温冰箱。文献上草甘膦的检测大部分要用衍生化。随着LC-MS/MS的推广,已经有专家研究在LC-MS/MS上不用衍生化检测草甘膦的方法。我们今年也购进了AB的Triple Quad 4500。我们想利用手中的这一利器,研究不用衍生化检测茶叶中草甘膦的方法。不知哪位同行有相关的资料?
按NY/T1096-2006《食品中草甘膦残留量测定》草甘膦衍生时一定要经过干冰浴(-40- -60摄氏度)吗?是否可以绕过此步骤?原理是什么?
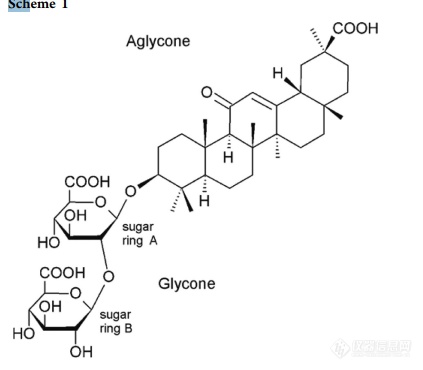
[align=center]CNS食品添加剂—甘草酸盐性质概述[/align] 杨勉疾[align=center]2021年 7 月[/align]1.甘草酸盐系列物质理化性质概述1.1 甘草酸理化性质 甘草入药史自古以来,是最为广泛的药用植物之一。其中甘草酸(CA)被认为是其提取物中最主要活性成分。甘草酸呈白色结晶性粉末,甜度约为蔗糖的200倍。显甜迟后,但留甜时间长;相对密度(d204):1.43;熔点在212-217℃左右;常压沸点972℃;闪点288℃;溶解性:难溶于冷水,易溶于热水,不溶于油脂,其热水溶液冷却后呈黏稠冻胶状。溶于丙二醇。 GA是一种单桥皂甙,其由三萜类疏水性苷元(18β甘草次酸)与亲水性二葡萄糖醛酸结合而成,GA的两亲性结构决定了其性能溶液中的物理性质。使得GA分子聚集水溶液中的表面活性化合物会导致聚集体、胶束的形成,并且在较高浓度下尤甚。其皂苷结构决定了GA许多特殊药理功能,调节其疏水分子形成水溶性复合物能力,可以用于调节其他物质化学稳定性,水溶性,生物利用度;以及在临床上应用于能性药物释放系统(DDS)。其有急性毒性:人体口经TDLo:280mg/kg/4W;小鼠口经LCLo:3gm/kg;小鼠腹经LCLo:2gm/kg;小鼠静脉LC:300mg/kg。在环境方面,甘草酸对水稍有危害,不可使未稀释或大量的产品接触地下水、水道或者污水系统。若无政府许可,不得排入周围环境。[1] 下图1.2分别为二维糖平台与三萜组成的基本结构单元透视图从两边伸出的部分;球和棍子(b)和空间填充(c)表示,显示由相互渗透的基本元素形成的通道单位(以浅灰色和深灰色显示的分子属于相邻单位)。通道约占晶体体积的42%。[align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108081544564157_4482_1608728_3.png[/img][/align][align=center]图 1甘草酸二维结构[size=16px][2][/size][/align][align=center][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108081544567370_8_1608728_3.png[/img][/align][align=center]图 2 甘草酸三维立体结构[2][/align] 甘草酸作为一种多元酸,在碱性或离子液体内会不同程度脱质子成盐,在自然条件下,会和钾、钠离子结合存在。甘草酸盐是由甘草酸衍生的一系列盐类总称,包括甘草酸铵、甘草酸一钾及三钾、甘草甜素二钠等。1.2 甘草酸铵 甘草酸铵为白色粉末或淡黄色结晶型粉末,有强甜味,甜度约为蔗糖的200倍,溶于氨水,不溶于冰乙酸。应用于甜味剂,依照我国《食品添加剂使用卫生标准》,可按生产需要适量用于肉类罐头、调味品、糖果、饼干、蜜饯凉果、饮料等等。还可以用于进一步制备其他甘草酸盐类的中间物。 甘草酸单铵盐具激素样活性,但无激素的副作用,不仅对气管炎、支气管炎、咳嗽、哮喘等呼吸系统疾病有显著疗效。而且对消化道感染、乙肝、口腔溃疡、胃溃疡等也有奇效。对于多种毒素如白喉毒素、河月豕毒素、破伤风毒素和蛇毒等有着较强的解毒功效。同时还具有类似肾上腺皮质激素的作用。其毒理学半数致死量为10g/kg;经骨髓微核实验证实无致突变作用[3]。1.3 甘草酸一钾及三钾 甘草酸一钾及三钾类似白色或淡黄色粉末,无臭。有特殊甜味(甘草酸一钾为蔗糖的500倍;甘草酸三钾为蔗糖的150倍),甜味残留时间长,易溶于水,溶于稀乙醇、甘油、丙二醇,微溶于无水乙醇和乙醚。其同样应用于甜味剂,和甘草酸铵类似;毒理依据其半致死量为小鼠口服>10g/kg[4]。 在化妆品行业,可配制成护肤霜,祛斑霜高级珍珠膏等,既有美容护肤,又能消炎、抗变态反应,治疗皮肤病等作用;在医药行业,可用于眼药水、口腔炎的药膏;在日化行业,可用于牙膏。1.4 甘草酸二钠 甘草酸二钠又名甘草甜素二钠。为白色至淡黄色粉末,味极甜,稀释4000倍仍有甜味,甜度约为蔗糖的150-200倍,且甜味残留时间长。易溶于水,溶于稀乙醇、甘油、丙二醇,不溶于无水乙醇、乙醚、氯仿和油脂。用作甜味剂。日本限用于酱油(0.015g/L)和豆酱(0.03-0.07g/L)。毒性为半致死量5g/kg[5]。 由于其在水中非常易溶解,溶液澄清透明,无杂质和怪味,口感好,在食品添加剂方面具有低热能、安全无毒和较强的医疗保健功效,是高血压、肥胖症、糖尿病、心脏病患者使用的最理想甜味剂,有浓郁的甘草特殊香味,具有保健、解毒、护肝、消炎、增香等功效,是非常理想的纯天然甜味剂原料。2.甘草酸盐的制备及检测标准2.1 甘草酸生产方法及指标[6] 甘草酸以甘草为直接制备原料。将甘草的根茎干燥后粉碎至0.833mm的粉末(保留纤维部分)取粉末及纤维200kg,加水1200kg,在85-100℃下浸提2h。过滤后滤渣再用1000kg水提取2h,过滤后滤渣再重复浸提1次。合并3次滤液,在搪瓷蒸发器中浓缩至1/5体积。冷却后加入95%乙醇,使乙醇浓度达到65%,静置24h,过滤除去植物蛋白、多糖等杂质。滤液中加入硫酸,调节PH至甘草酸沉淀析出。过滤。洗涤后,加入3倍的丙酮,加热可回流3h,倾出提取液,残渣再反复回流提取2次。合并3次提取液,过滤后回收丙酮,浸湿甘草酸,与45℃干燥1h,缓缓升温至85-95℃,快烘干时,升至100-105℃烘干5min,经粉碎后即得成品。 此外,也可直接用氨水萃取,经浓缩后用硫酸沉淀,再用95%乙醇重结晶而得。 其质量指标需要符合中国企标:水分≤13%;灰分15%;熔点为220℃。2.2甘草酸二钠制备及质量标准[7] 甘草酸二钠一般由甘草酸为直接原料。其一由甘草甜素与钠碱进行部分中和而后精制而成。其二,由甘草粉加五倍水煮沸抽提,滤去固形物,加稀硫酸至呈弱酸性。室温下放置至析出物沉降,除去上澄清液,沉淀经水析出后用氨水中和、过滤、滤液加醋酸使甘草甜素铵析出,用70%-80%乙醇重结晶,按理论值加入碳酸钠水溶液,减压浓缩而得。 其质量指标参照日本标准,1999。包括含量95%-100%,溶液性状:10%水溶液应透明;5%溶液PH值5.5-6.5,氯化物(Cl-计)≤0.014%;水分≤13%;砷含量<4mg/kg;重金属<40mg/kg等等。相应的质量指标分析手段一般均通过标准试剂化学滴定得到。2.3甘草酸一钾及三钾制备方法及质量标准[8] 以甘草酸粗品(含量75%)为原料,在乙醇中用氢氧化钾中和而得。将100g甘草酸盐粗品加入400ml工业乙醇中国,在40-50℃下搅拌提取1h。抽滤后滤渣用200ml乙醇在同样的条件下提取1h,合并提取液,在搅拌下加入20%的KOH乙醇溶液至PH至7-8为止。静置片刻后分离得甘草酸三钾黄色结晶200g,将其放入80-90ml冰醋酸中,加热至75℃,保温几分钟使其转化为单钾盐,抽滤得近白色甘草酸单钾盐粗品,用少量工业乙醇洗涤一次,以出去黄酮类色素和甘草次酸等杂质。粗品用400ml乙醇冰醋酸混合液溶解,加入10g活性炭,在80℃下脱色0.5h。过滤后滤液放置结晶,得产品25-30g,收率约为70%。 其质量指标包括含量(UV法≥98%;HPLC≥85%);重金属≤0.001;砷盐≤0.0002;灰分≤9.63%;水中不溶物≤0.5%。2.4 间接甘草酸盐生产制备方法 为使甘草酸发挥更好的疗效和提高生产效率,非常需要实用性较强的制备甘草酸盐精品方法。 根据甘草酸易溶于热水,可溶于热稀醇,几乎不溶于无水乙醇和乙醚, 又可于水溶液中加稀酸游离液,又可于水溶液中加稀酸游离出来的性质,以及甘草酸锌盐、铁盐、铝盐及秘盐在热水中仅微溶或者不溶的性质,可以使甘草酸在水或稀醇溶液中与相应的无机盐水溶液反应制取需要的甘草酸盐。如果选用粗甘草酸溶液作原料,则得到甘草酸盐粗品,要制成精品往往需要反复多次精制,[font=times new roman][size=13px] [/size][/font]操作十分繁琐.如果选用甘草酸单按盐精品为原料,[font=times new roman][size=13px] [/size][/font]可以比较方便地制取草酸盐精品。在实际生产中,可以利用甘草或者甘草浸膏为原料,先制取甘草酸单按盐精品,然后再以甘草酸单按盐为原料制备甘草酸盐别的品种。在质量指标检测方面,甘草酸根含量测定可采用层析法,锌、秘、铝和铁的测定可采用容量分析或重量分析的方法。2.4.1甘草锌制备 取甘草酸单铁盐209溶于80%乙醇90ml中,加热回流,慢慢滴加予热至50℃的5%硫酸锌溶液80g,生成白色沉淀,加完硫酸锌溶液后,保温反应30min,之后降温至20℃,过滤,滤饼用6oml蒸馏水分三次清洗,滤尽母液,取出滤饼真空50℃干燥,得棕黄色甘草锌粉末19.69。测定甘草酸根含量87.6%,锌含为10.5%。2.4.2甘草酸秘制备 取甘草酸单铵盐溶于200ml热水中,于8℃在搅拌下慢慢滴加予热至60℃的10%的硝酸秘酸性溶509,需维持反应液为酸性(PH~3),生成白色沉淀,加完硝酸秘溶液后,保温反应30min,然后降温至30℃,过滤,滤饼用60rnl蒸馏水分三次清洗,滤尽母液,再以95%乙醇45ml分三次清洗,滤尽母液,在40~50℃真空干操,得白色甘草酸秘粉末21.39,测定甘草酸根含量82.2%,秘含量14%。3.甘草酸盐应用 邓淑华等人研究显示,甘草酸二钠、甘草酸二钾、甘草酸二铵在体外实验条件下,对金黄色葡萄球菌、白色葡萄球菌、大肠埃希氏菌、福氏志贺氏菌、乙型副伤寒沙门氏菌等细菌均表现了不同程度的抑菌作用。实验额外证实,甘草酸盐对乙型副伤寒沙门氏菌、金黄色葡萄球菌(附院)、福氏志贺氏菌等细菌具有一定的杀菌作用[9]。 甘草酸盐及甘草煎剂对杀虫双染毒的小鼠急性中毒不仅有顶防作用,而且甘草酸盐对急性中毒还有治疗作用,能明显降低杀虫双不同途径染毒之小鼠 、兔子的死亡率、其解毒机尚待进一步研究[10]。Francesco Maione[font=宋体]等人对单铵甘草酸盐抗炎抗伤害以小鼠实验进行以及生化和对接研究。在小鼠单次给药后的,一次腹腔注射AG对酵母多糖引起的足跖水肿和足跖肿胀均有抗炎作用腹膜炎。此外,在几种疼痛动物模型中,如扭体试验、福尔马林试验,酵母多糖诱发的痛觉过敏,试验前24小时给予AG可诱发痛觉过敏强烈的抗伤害作用。综上所述,所有这些发现都突出了AG在疼痛和或炎症相关疾病临床治疗中的潜在应用。AG与mPGES-2和COX-2的关键氨基酸相互作用。经过实验结果分析,甘草酸单铵的抗炎抗伤效应来自其与mPGES-2和COX-2的特异受体相互作用 。AG在结合处的定位较好COX-2与Trp387、Ser530(氢键)和Arg120等关键氨基酸相互作用时的囊袋。此外,通过结合刚性和柔性分子对接研究,两种可能的方法提出了AG与5-LO相互作用的机制:非氧化还原竞争结合和非氧化还原竞争结合Fe[/font][font=宋体]2+[/font][font=宋体]络合。而理论计算结果显示,前者结合能相对更低。[/font][font=宋体][11][/font]Carlotta Marianecci等人[font=宋体]研究表明甘草提取物可用于治疗皮炎、湿疹和银屑病,其疗效与皮质类固醇相当。在这项工作中,通过研究不同浓度的表面活性剂(吐温85和司班20)和胆固醇组成的囊泡在甘草酸铵(AG)释放中用于治疗各种炎症性疾病的效果。对囊泡进行了包括尺寸、ζ电位、各向异性、药物包封率、稳定性、细胞毒性评价和皮肤耐受性等方面的表征,证实纤维素膜在甘草酸铵囊泡的体外释药特性中作用[/font][font=宋体][12][/font][font=宋体]。[/font]甘草酸在大多数肝脏疾病的临床实践中用作肝脏保护剂。万荣等研究证实,甘草酸二铵减缓肝损伤并可阻止自然杀伤T细胞。其通过两种不同剂量甘草酸多铵给药对照试验,通过检测相应指标。得出预处理能显著降低血清ALT并改善cona诱导的自身免疫性肝组织损伤的结论。实验结果证实,DG预处理可下调攻击后的炎性细胞因子与Con A,并可以抑制胸腺T淋巴细胞凋亡。此外,甘草酸二铵还可有效地抑制CD4的增殖+CD25、CD69+、CD8+及CD69型+等外周血和脾脏的亚群,并显著下调NKT细胞的频率,同时上调树突状细胞的频率肝脏[13]。隋秀文等研究证明了甘草酸多铵盐和氯化锂共同作用抑制伪狂犬病病毒PrV感染,并可诱导PrV细胞凋亡。(PrV)是一种猪嗜神经性疱疹病毒与单纯疱疹病毒1型(HSV-1)有共同的基因组排列。其感染严重威胁畜牧业和人类健康。以甘草酸多铵盐为基底开发有效的抗病毒药物是减少PrV感染的重要策略之一[14]。李云等研究证实,甘草酸二铵(DG)具有抗炎和保肝药理作用。非酒精性脂肪肝(NAFLD),作为常见的慢性肝病,在世界范围内普遍存在。李云团队通过高脂饮食诱导的NAFLD模型小鼠实验,我们观察到DG可以减轻体重、肝脏脂肪变性以及肝脏炎症Illumina对16S rRNA的测序显示DG干预改变NAFLD小鼠肠道微生物群的组成,使得肠道菌群的丰富度显著增加。特别是DG降低了厚壁菌与拟杆菌的比率和产生内毒素的细菌(如脱硫弧菌)提高了益生菌如变形杆菌和乳酸杆菌的丰度。DG能增强短链蛋白的表达水平,如产脂肪酸(SCFA)的细菌、瘤胃科和漆树科,促进SCFA的产生。此外DG补充显著减轻了肠道低度炎症。促进细胞表达紧密连接蛋白、杯状细胞数量和粘蛋白分泌,从而增强肠屏障功能。因此,目前可以认为,DG对NAFLD的预防可能是通过调节肠道菌群和恢复肠道功能来实现的[15]。异甘草酸镁(MgIG)被广泛应用于慢性肝病的治疗。主要认为是通过作用于肝毒性诱导物质——甲氨蝶呤(MTX)诱导的肝毒性实现其效果。曹雨竹等人研究结果显示,预防性的给予小鼠MgIG(9和18mg/kg/天)可显著降低小鼠血液中血清天冬氨酸转氨酶和丙氨酸转氨酶的减少;MgIG还能减轻MTX诱导的肝纤维化。对MTX诱导的肝细胞损伤有较好的保护作用。此外,MTX还可诱导环氧合酶-2(COX-2)表达,给予MgIG后,肠道通透性和炎症减轻。总之,MgIG对甲氨蝶呤引起的肝毒性和肠道损伤有积极作用一种,是有可能缓解MTX肝脏和肠道副作用的药物[16]。4.总结甘草是一种豆科草本植物,其作史古已有之,必然意味着甘草所独具的 性质千百年来一直为人们所使用。而其主要活性成分甘草酸及其衍生盐类由于其甜度极高,且甜度留存时间长,主要用作甜味剂用于食品添加剂中。但都具有一定毒性,需要严格按照国家标准使用。此外,甘草酸盐还具有药理性质,在生物医药研究方面受到了学者的广泛关注,具有抗炎、保肝两方面的功能,因此也频繁应用与新型药物的开发,其价值也得到了更多的延伸。参考文献[1]甘草酸的制备及其在食品工业中的应用.食品工业,1994,(6);49~51[2]Tykarska E , Gdaniec M . Toward Better Understanding of Isomorphism of Glycyrrhizic Acid and Its Mono- and Dibasic Salts[J]. Crystal Growth & Design, 2013, 13(3):1301-1308.[3]郑国斌.从甘草酸粗品制取甘草酸单钾盐.中国医药工业杂志,1995,26(2);54[4-5,7-8]食品添加剂应用手册/孙平,张津凤主编.一北京:化学工业出版社,2010.10 ISBN978-7-122-09417-9[6]苌云玉.甘草酸盐制备方法研究[J].基层中药杂志,1995(04):33-34. [9]邓淑华,王晓斌,王鸿梅,刘艳华.甘草酸盐抗菌作用的实验研究[J].承德医 学院学报,2011,28(03):325-326.[10]黄能慧,曾样锬,刘季昆,夏炳南.甘草酸盐对农药(杀虫双)的解救作用[J].贵阳医学院学报,1982(03):21-22.[size=13px] [/size][11] Maione F , Minosi P , Giannuario A D , et al. Long-Lasting Anti-Inflammatory and Antinociceptive Effects of Acute Ammonium Glycyrrhizinate Administration: Pharmacological, Biochemical, and Docking Studies[J]. Molecules, 2019, 24(13)[12] [size=13px][color=#222222]Koide M , Takahashi M , Tamagaki S , et al. Catalytic effect of dipotassium glycyrrhizinate on the hydrolysis of nonionic ester surfactants[J]. Journal of the American Oil Chemists' Society, 1996, 73.[/color][/size][13]万荣, 刘莎, 范稚坚,等. Clinical Observation of Diammonium Glycyrrhizinate Enteric-coated Capsule in Preventing Liver Injury Induced by Anti-tuberculosis Drugs[J]. 大理学院学报, 2019, 004(004):45-47.[color=#222222][14] Sui X , Yin J , Ren X . Antiviral effect of diammonium glycyrrhizinate and lithium chloride on cell infection by pseudorabies herpesvirus.[J]. Antiviral Research, 2010, 85(2):346-353. [15][/color] [color=#222222]Li, Yun, Liu, et al. Diammonium Glycyrrhizinate Protects against Nonalcoholic Fatty Liver Disease in Mice through Modulation of Gut Microbiota and Restoration of Intestinal Barrier[J]. Molecular pharmaceutics, 2018.[/color][16] Marianecci C , Rinaldi F , Mastriota M , et al. Anti-inflammatory activity of novel ammonium glycyrrhizinate/niosomes delivery system: Human and murine models[J]. Journal of Controlled Release, 2012, 164(1):17-25.
草甘膦产品按SN/T 1923-2007处理衍生后进行调谐,为什么草甘膦和草甘膦内标在调谐过程中都是在母离子397左右有很高的响应,标准上草甘膦是392.1的母离子,草甘膦内标是395。
为什么草甘膦按国标做不稳,流动相增加对应盐,却稳了
根据国标《GBT 5750.9-2006 生活饮用水标准检验方法 农药指标》测水中的草甘膦和呋喃丹,用柱后衍生法,需要用到邻苯二甲醛(OPA)和2-巯基乙醇做衍生试剂,不知道这两种试剂在哪里能买到呀,问了几家卖试剂的,都没有,这两种是不是不属于常规试剂啊,有谁知道哪能买到呢,试剂的规格还有大概价格是多少呀,急用,多谢了!
公司要求检测草甘膦,但现在连离子对都找不到,尝试过用SN/T 1923-2007上的方法,也试过提高衍生试剂的浓度,更换流动相,或者提高曲线的浓度,有没有哪位社友用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测草甘膦成功的?希望各位社友传授点经验

10,抽取5个版友);中奖名单:捌道巴拉巴巴巴(注册ID:v3082413)莫名其妙(注册ID:moyueqiu)翠湖园(注册ID:hhx050)dahua1981(注册ID:dahua1981)玲儿响叮当(注册ID:jshbhh)http://ng1.17img.cn/bbsfiles/images/2017/02/201702081513_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/02/201702081513_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================RP-HPLC法同时测定复方甘草酸苷片中三组分的含量方法:HPLC基质:药品应用编号:102819化合物:甘草酸单铵盐, 甘氨酸, 蛋氨酸固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil C18(2) 5u 150 x 4.6mm色谱条件:色谱柱:Diamonsil C18(2) 150×4.6 mm, 5μm(Cat#:99601) 流动相:乙腈-0.01 mol/L磷酸溶液(40:60) 流速:1.0mL/min 进样量:20μL 检测器:UV 257nm,262nm文章出处:中国药物应用与监测 2009, 3(6):153-155关键字:方甘草酸苷片, 甘草酸单铵盐, 甘氨酸, 蛋氨酸, 高效液相色谱法, HPLC, 钻石二代, Diamonsil C18(2)谱图:摘要 目的:建立高效液相色谱法同时测定复方甘草酸苷片中甘草酸单铵盐,甘氨酸和蛋氨酸的含量.方法:采用Diamonsil C18色谱柱(150×4.6mm, 5μm),流动相为乙腈-0.01mol/L磷酸溶液(40:60),流速1.0mL/min,甘草酸单铵的检测波长为257nm,甘氨酸和蛋氨酸的检测波长为262nm.结果:甘草酸单铵盐,甘氨酸和蛋氨酸浓度分别在50.52~505.20μg/mL,10.6~106.0μg/mL,10.4~104.0μg/mL范围内呈良好的线性关系;平均回收率分别为(100.32±1.35)%,(101.02±1.80)%和(100.75±1.86)%,RSD分别为1.35%,1.78%和1.85%(n=9).结论:本法可用于同时测定复方甘草酸苷片中甘草酸单铵盐、甘氨酸和蛋氨酸的含量,方法简便,结果准确.图谱:http://www.dikma.com.cn/Public/Uploads/images/71_7_1.jpghttp://www.dikma.com.cn/Public/Uploads/images/71_7_2.jpg
哪位大侠做过草甘膦?GCMS法,为什么我最后只能出它的衍生物的峰,草甘膦的峰怎么不出呢?我把浓度都加到120ppm了,还是不出峰,倒是它的衍生物出峰很稳定,这究竟是为什么啊?
最近在做草甘膦,主要是参照SN /T 1923-2007进出口食品中草甘膦残留量的检测方法LCMSMS法,依照标准衍生,标准溶液中加入硼酸和衍生试剂FMOC-CL,上质谱,正离子模式,根本找不到392目离子,衍生试剂的浓度增加了,也没有作用,不知道问题出在哪里了?很是头痛!那位高人做过,请指教!十分感谢!很着急啊做不出来。希望做过的前辈指教。
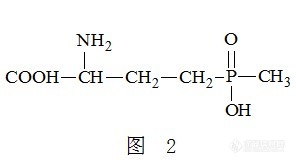
关于”新版GB 2763 食品安全国家标准 规定茶叶中限量农残草甘膦和草铵膦项目“的检测研究 一、研究意义及现状 随着新版GB 2763 食品安全国家标准的不断更新及发布实施,草甘膦和草铵膦已被明确列为茶叶中农药残留强检(必检)项目,草甘膦在茶叶中的限量为1mg/kg,草铵膦在茶叶中的限量为0.5mg/kg。同时,草甘膦和草铵膦也成为中国茶叶出口国外的检测项目(来源于中华人民共和国商务部),且已成为越来越严的限量指标。 文献(2013年农药行业预测和草甘膦市场机遇分析,杨益军,农药市场信息,2013.03)报道,除草剂草甘膦因其高效、广谱、低毒等特性使其被广泛应用,未来需求量也将大幅增加。但草甘膦的使用容易使植物产生抗性(IARC国际研究机构发布报告称草甘膦很可能对人类致癌),而草铵膦可克服该缺陷,现已有学者(草铵膦、百草枯、草甘膦对非耕地杂草的防效比较,凌进,农药,2014年第53卷第8期,613-615)对草铵膦和草甘膦的除草性能进行了研究,确证了草铵膦代替草甘膦的可行性。 因草甘膦和草铵膦为广谱除草剂,被广泛应用于农业、林业及园艺的栽培。我国作为农业大国,其茶叶产量世界第一、出口量世界第二,草甘膦和草铵膦的生产和使用量都位居世界前列(草甘膦 草铵膦及其代谢产物的检测方法,李小娟、周信康、孟品佳,公共安全中的化学问题研究进展)。同时,我单位对西南茶叶原料主产区进行了初步调研,进一步确认茶农使用草甘膦和草铵膦农药的现状。 随着草甘膦和草铵膦除草剂使用量的日益增大,使其常被发现存在于环境水样、土壤及植物中,这样长期积累会引起环境污染,从而对人类健康造成严重威胁。草甘膦和草铵膦结构类似,且均含有膦酸基、羟基、氨基,是极强的两性化合物,易溶于水,难挥发。鉴于草甘膦和草铵膦特殊的物化性质和茶叶基质自身的复杂性,无论国内外,茶叶中草甘膦和草铵膦同时检测的标准还未见发布。 目前,可用于检测草甘膦和草铵膦农药残留量的主要方法有液相色谱法,柱前衍生后气相色谱法、气相色谱-质谱法及液相色谱-质谱/质谱法。 快速发展起来的超高效液相色谱-质谱联用技术,具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,当今已成为检测型实验室检测农残的首选。然而,若采用液质质直接测定草甘膦和草铵膦,则仪器响应较低,无法满足茶叶中草甘膦和草铵膦农药残留量检测的要求。 近两年来,已有研究文献陆续发表,用柱前衍生-液相色谱串联质谱法检测。笔者结合其文献研究结果,对茶叶中草甘膦和草铵膦农药残留量的检测方法系统地进行研究,采用9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂,在硼酸盐缓冲盐溶液的条件下,能与草甘膦和草铵膦的提取液发生衍生反应,形成衍生产物,衍生产物注入UPLC进行色谱洗脱分离,采用串联质谱探测响应信号,外标法直接快速定量茶叶中的草甘膦和草铵膦的含量。二、液质质检测分析原理 质谱原理是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。液质联用是将色谱的分离能力与质谱强大的定性功能结合起来,实现对复杂混合物更准确的定量和定性分析,简化样品的前处理流程,使样品分析更简便。主要针对不挥发性、极性、热不稳定、大分子量等化合物的分析测定。液质联用检测技术灵敏度高,且串联质谱(三重四级杆)定性准确,可有效杜绝微量甚至痕量物质分析时的假阳性现象,常用于目标物质的痕量分析。 采用柱前衍生-液相色谱串联质谱法检测茶叶中的草甘膦和草铵膦有以下优势: 1)、灵敏度高、线性好、检出限低(可达ng/mL级及其以下); 2)、定量结果准确、稳定、重复性好; 3)、实验操作简单、步骤少、耗时短、分析速度快、检测效率高; 4)、实验试剂无污染、无毒、安全; 5)、有效减弱基质对目标物检测的影响。三、茶叶中草甘膦和草铵膦农药残留量检测的前处理试验 茶叶经GB/T8303磨碎、过筛制得待测茶样(发酵茶应先低温去除水分,使样品易于磨碎); 准确称取已磨碎处理过的茶样1g(精确至0.001g)置于80mL具盖离心管中,加入10mL水,涡旋混匀静置,加入2mL二氯甲烷,混匀,超声提取或回旋振荡10min,低速离心机4500r/min离心5min,取上清液,制得提取上清液; 注意:若茶样为新采摘的鲜叶,则称取约5g鲜叶于研钵中,加入30mL水,研磨约10min,将其转入离心管中,用10mL水洗涤研钵后转移至离心管,重复洗涤一次,再次加入10mL二氯甲烷于离心管,均质至混匀,4500r/min离心10min,取上清液,制得提取上清液; 将提取上清液用净化柱CAX、C18,以及活性炭小柱等进行比对试验,确定以C18小柱净化提取液,制得提取净化液; 通过缓冲液浓度、衍生液浓度、衍生液用量、缓冲液用量、净化液用量,衍生时间等条件试验,得出最优衍生试验参数为缓冲液浓度为50g/L,衍生液浓度20g/L,衍生液用量:缓冲液用量:净化液用量的体积比为1:1:1,衍生时间为约3h,衍生液过0.22μm的有机滤膜后进样。四、茶叶中草甘膦和草铵膦农药残留量检测的衍生机理 茶叶中草甘膦和草铵膦农药残留量的衍生机理为:在硼酸钠缓冲盐溶液条件下,草甘膦(分子结构如图1所示)和草铵膦(分子结构如图2所示)中R-NH-R’的-H被FMOC-Cl(分子结构如图3所示)中的FMOC-取代,生成 http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414494663_01_0_3.png,得到衍生目标产物草甘膦衍生物和草铵膦衍生物。 其中,草甘膦分子结构图,见图1;草铵膦分子结构图,见图2;9-芴甲氧羰酰氯(FMOC-Cl)分子结构图,见图3;草甘膦和草铵膦与9-芴甲氧羰酰氯(FMOC-Cl)的衍生机理图,见图4。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414424276_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414430242_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414432007_01_2275853_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507061123_553629_2275853_3.png五、茶叶中草甘膦、草铵膦农残衍生物在质谱中的裂解机理1、茶叶中草甘膦农残衍生物在质谱中的裂解机理 通过对草甘膦衍生物在串联质谱中的裂解机理进行系统的分析研究,可探索出草甘膦衍生物的裂解机理为:首先草甘膦衍生物裂解为离
单位的Thermo的TSQ[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]刚装好使用1个多月,本人以前从未做过[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]和农残,最近要做草甘膦及代谢物,按照SN/T1923-2007标准,配标液,衍生化,结果到质谱调试时,找不到分子离子峰,用的条件是:ESI 正离子 3500V 350度 用1ppm浓度标样进行质谱条件优化,可是m/z392和m/z334强度只有4次方,而其他离子相应很高:336,363,635,这几个离子进草甘膦单标和草甘膦代谢物单标时都会出现,强度都有6次方,,纳闷,想不出问题出在哪?会不会是衍生化不完全,记得加入FMOC-CL时溶液白色浑浊,过夜后就澄清了,应该反应完全了吧流动相是0.1%甲酸-2mmol/l乙酸铵+乙腈+甲醇
1实验目的本文对茶叶中的草甘膦残留经提取以及WAX和MCX固相萃取柱的净化,应用HPLC-QQQ进行定量分析,通过重复性、线性关系和回收率等参数进行考察,所建立的方法能对草甘膦进行快速、高灵敏的检测分析。1 材料与方法1.1材料和试剂草甘膦标准品,甲醇(色谱纯),氨水(优级纯), 原乙酸三甲酯:色谱纯,实验用水均为超纯水。1.2仪器设备 液相色谱-三重四级杆质谱联用仪:Agilent 1290-6460、MCX固相萃取柱:150mg 6mL,CNW、 PWAX固相萃取柱:150mg 6mL,CNW、恒温水浴锅;1.3 标准品配制 将草甘膦标准品用50%甲醇/水分别配制成50μg/kg、100μg/kg、200μg/kg、500μg/kg、1000μg/kg的标准曲线,根据样品反应的流程进行反应。1.4 样品处理称取已粉碎好的茶叶试样2.5g置于离心管中,添加标准中间液,10mL二氯甲烷,水25mL,均质,水5mL清洗刀头,抽滤,水10mL提取残渣。5等分滤液,待净化。MCX和PWAX(150mg/6mL)依次用5 mL甲醇、5mL水平衡,将平衡后的MCX与PWAX上下串联,将3.1的样液加载至MCX串联柱上,流出物弃去;5mL 水淋洗柱,弃去,弃去MCX柱,用5 mL甲醇淋洗PWAX柱,减压抽干3min,用10mL 5%氨水/甲醇洗脱接收,40-45℃减压蒸干。加入0.5mL乙酸溶解残渣,转入到15mL离心管中,加入2mL原乙酸三甲酯溶解残渣,合并溶液,涡旋1min,100℃加热2小时进行甲基衍生化反应,转移到50mL旋转蒸发瓶中,甲醇洗离心管合并到50mL旋转蒸发瓶中,50℃减压浓缩至干,1mL 50%甲醇/水定容,待测。1.5仪器条件1.5.1色谱条件a. 色谱柱:Agilent Poroshell 120 EC-C18 3.0X100mm,2.7-Micron; b. 流动相及其梯度流速:0.4mL/min时间(min)0.1%甲酸+2mMol/L乙酸铵水溶液(%)甲醇(%)0.009551.009556.0010908.0010909.0095510.009551.5.2 质谱条件a) 离子源:ESI源;b)扫描方式:正离子扫描;c)检测方式:多反应监测;e)电喷雾电压:4000V;d)雾化气温度:350℃;f)雾化气流速:10L/min;g)雾化气压力:40psi;h)定性离子对、定量离子对、提取电压以及碰撞气能量等见表。名称Q1Q3提取电压 V碰撞能量 V草甘膦254152951025410295102547495204.实验结果将草甘膦分别反应浓度为50μg/kg、100μg/kg、200μg/kg、500μg/kg、1000μg/kg的标准曲线,以峰面积y为纵坐标,浓度x为横坐标,绘制标准工作曲线方程为:y=3037.390732x+26813.423347,相关系数R2=0.99969630。茶叶样品中草甘膦的浓度为0μg/kg,添加浓度为100μg/kg,添加回收浓度为76.325μg/kg,回收率为76%。
各位好,我是做水质的,用Waters的液质做草甘膦、丙烯酰胺及三氯酚,尽管调谐成功了,可做的效果一点也不好,峰的干扰太大了,谁有相关的一些仪器参数,供参考一下。
小弟是个刚开始做液相的新手,做的是一个季铵盐,N+在两个环之间,位阻很大的样子,正在尝试往流动相里面加入离子对的方法以下是一些实验结果1.流动相为乙腈-水=20:80,季铵盐和水的保留时间均为1.3min2.流动相比例不变,水相加入15mmol/L的SDS,季铵盐的保留时间不变3.流动相比例不变,水相加入10mmol/L的NaClO4,季铵盐的保留时间增加到2.2min我猜测是SDS结合能力比CLO4-要低一些我想问的是,三氟乙酸的结合能力和CLO4-的结合能力哪个强,另外还有什么可用的离子对试剂另外参照了药典05版的酸性染料比色法,加入溴麝香草酚蓝,然后用氯仿萃取,氯仿层用紫外测,线性关系非常好,氯仿层进样,用液相却没有出现任何结果(10%-100%乙腈50分钟,然后100%乙腈10分钟)酸性染料比色法也没有查到任何用HPLC的文献,不知这是为什么谢谢大家bow~~~
41%草甘膦异丙胺盐水剂生产中的几个主要问题精细化工中间体01.5.9 杨晓玲谢谢各位
有谁有草甘瞵异丙胺盐水剂的检测方法呀,我急需其方法及注意事项。谢谢各位了!
请教下做水中草甘膦的同行,不知道大家做的怎么样。我们用柱后衍生来做没装机的时候做的很好,但是到自己做的时候就不行了,主要是响应值低了好多好多。不知道大家有没有出现过这样的问题。另外做完草甘膦之后,柱子是怎么维护,什么时候用什么溶剂冲洗,我都绕晕了。

农药残留草甘膦的检测前言 草甘膦又叫镇草宁,是广谱除草剂,对哺乳动物有低毒性。除草剂的广泛使用使得草甘膦成为地下水甚至饮用水的重要污染物。简介 分析草甘膦的方法现在主要有气相色谱法或液相色谱法,通常都要衍生化处理,其中采用液相色谱法更多些。现主要介绍比较复杂的阳离子交换柱后衍生荧光检测器测定草甘膦的方法。本方法着重描述了分析草甘膦及其主要代谢物氨甲基磷酸(AMPA)的便利方法。 本方法参考了美国EPA方法、GBT 5750.9-2006方法、岛津、美瑞泰克、博纳艾杰尔等公司提供的参考方法及本实验室全体工作人员的集体智慧编制而成。阳离子交换柱后衍生荧光检测器法测定草甘膦色谱原理 柱后衍生与荧光检测器结合使得本方法具有很高的灵敏度和选择性。样品进样后,草甘膦和AMPA经流动相磷酸二氢钾缓冲液洗脱,在色谱柱中分离。分离后,分析物通过柱后衍生反应系统,生成的反应产物用荧光检测。由于每个厂家的仪器的灵敏度各不相同,所以每种方法的进样量也不相同,一般为10-200ul.此方法建议定量进样50uL,如图1所示,效果很好。http://ng1.17img.cn/bbsfiles/images/2013/09/201309200103_465474_2369266_3.png图11.草甘膦 1.0mg/L 2.AMPA 1.0mg/L设备 L600高效液相色谱仪,包括:梯度泵1套,柱后衍生仪1台,衍生注射泵2台,荧光检测器1台,数据采集控制器1台,自动进样器1台,柱温箱1台,阳离子色谱柱及保护柱各1根,色谱工作站1套,超声波振动仪,溶剂过滤器,固相萃取装置,离心机。试剂 5 mM 磷酸二氢钾,磷酸调pH 2.0 ;5 mM 氢氧化钾 ;5%次氯酸钠溶液;硼酸钠缓冲稀释剂;OPA(二巯基乙醇),色谱级;甲醇,色谱级;硫醇试剂。试剂和标准品的准备氧化试剂(试剂1): 将一瓶次氯酸盐稀释剂倒入预先用甲醇清洗过的干净试剂瓶中。加入100uL5% 次氯酸盐溶液(家用漂白剂),旋转,混匀,0.45um水系滤膜过滤,超声脱气。加入的剂量可能需要根据检测器的响应进行调整:色谱系统平衡后,草甘膦混合测试溶液进样10uL,如图2所示。如果AMPA和草甘膦的面积比例差别很大,则在氧化剂里面再加入5%次氯酸盐溶液,每次加入20uL,直至二者所占面积比例相当。 OPA试剂(试剂2): 将OPA稀释剂倒入预先用甲醇清洗过的干净试剂瓶中。鼓泡10min以除去其中的氧气。余下的步骤应在尽可能短的时间内完成,因为此试剂对氧气和光敏感:称取大约100 mg OPA于一小烧杯中,加入10 mL甲醇溶解,而后加入OPA稀释剂。用1~2 mL甲醇清洗烧杯,把清洗液倒入稀释剂中。加入2 g硫醇试剂于试剂瓶中。0.45um有机系滤膜过滤,超声脱气,备用。色谱条件分析柱:草甘膦分析柱,阳离子交换柱,4 mm x 150 mm x 8um保护柱:草甘膦保护柱,阳离子交换柱,3 mm x 20 mm x 8um柱温:55 °C 淋洗液:(A)磷酸二氢钾(5 mM,磷酸调pH 2.0);(B)氢氧化钾(5 mM)流速:0.4 mL/min 柱后试剂1:氧化试剂,36 °C,流速为0.4 mL/min 柱后试剂2:OPA,室温,流速为0.4mL/min 荧光激发波长为330 nm,发射波长为460 nm 进样量:50ul剃度表:[fo
最近在做农药草甘膦的检测,发现进出口标准里SN/T 1923-2007 里,提取液过CAX柱,用13mLCAX洗脱液洗脱并收集,洗脱液45度旋转蒸发近干。(CAX洗脱液配方:160mL水+2.7mL盐酸+40mL甲醇)。实际在旋转蒸发过程中,半个小时感觉都没蒸发到多少?试过氮吹感觉也很难吹干,洗脱液含水量太大了。请问各位同行,是怎么处理的?
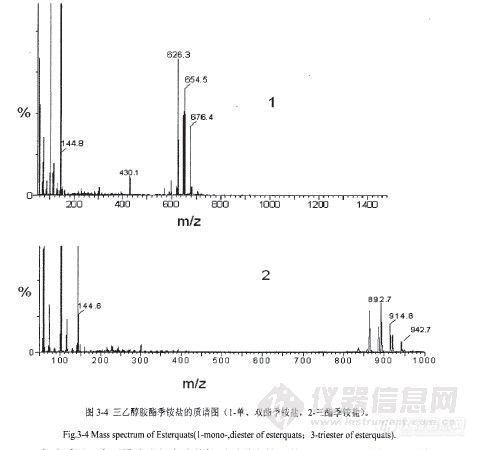
宫志鹏:建立了反相高效液相色谱法测定三乙醇胺酯中单、双、三酯含量的方法。采用Diamonsil C18色谱柱(250X4.6mm,5I,tm),柱温30。C,溶剂为氯仿,流动相为甲醇和氯仿,检测器为电雾式检测器。通过讨论k’值和兄与流动相极性、流速和柱温的关系,确定了梯度洗脱程序,30rain内三乙醇胺单、双、三酯得到了较好的分离。三乙醇胺单、双、三酯在20.500mg·L.1范围内线性关系良好,线性相关系数分别是0.9980、o.9991和0.9913,最低检出质量浓度分别(3s/N)是1.0mg·L~、1.5mg·L-1和1.5mg·L-1,相对标准偏差(n=6)分别是2.45%、1.81% 和1.98%。优化了反相高效液相色谱测定三乙醇胺酯季铵盐中单、双酯混合季铵盐与三酯季铵盐含量的方法。样品经过乙醚萃取处理,采用Inertsil CN.3色谱柱(250× 4.6ram,5Irtm),柱温50。C,溶剂为正丙醇,流动相为正丙醇和水,梯度洗脱, 检测器为电雾式检测器,并采用液相色谱.质谱法确证,15min内三乙醇胺单、双酯混合季铵盐与三酯季铵盐得到了较好的分离。单、双酯混合季铵盐与三酯季铵盐在25.750mg·L。1范围内线性关系良好,线性相关系数分别是O.9966和O.9913,检出F艮(3s/y)是0.8mg·LJ和10.0mg·L一,相对标准偏差(n=6)分别是0.5 l%和12.38%。:三乙醇胺酯,三乙醇胺酯季铵盐,梯度洗脱,高效液相色谱,电雾式检测器Abstract :An analytical method of reversed phase hi曲performance liquid chromatography(RP-HPLC)was developed for determination of triethanolamine mono-,di—and triesters.Established a gradient elution programme by analyzing the relationship of k’,R and elution rate,elution polarity,temperature. Triethanolamine mono-,di-and triesters were detected and separated successfully by Diamonsil Cls column(250x4.6mm,59m)and column temperature of 30"C,with chloroform as solvent and methanol-chloroform as the mobile phase in gradient elution and with a Charged aerosol deteror.The linear ranges of triethanolamine mono一,di-and triesters were 20-500 mg‘L。(r=O.9980),20-500 mg·L。1(r=0.9991)and 20—500mg‘L_(r=0.9913)respectively.The measurable lowest limits were 1.0rag‘L一, 1.5mg’L—and 1.5mg‘L—and the RSDs were 2.45%.1.8 1%and 1.98%respectively. An analytical method of reversed phase hi曲performance liquid chromatography(RP—HPLC)was optimized for determination of triesterquats in 1 5min.Mono-,diesterquats mixture and triesterquats were detected and separated ccessfully by Inertsil CN一3 column(250x4.6mm,5pm)and column temperature of 30。C.with n—propanol as solvent and n—propanol—water as the mobile phase in gradient elution and with a charged aerosol deteror.The linear ranges of mono一, diesterquats mixture and triesterquats were 25-750mg·L-1(FO.9966) and 25-750mg’L叫(r=0.9913)respectively.The measurable lowest limits were 0.8mg‘L。1 and 10.0mg。L-1 and theRSDs were 0.51%and 12.38%respectively. Key words:triethanolamine esters,esterquats,gradient elution,high performance liquid chromatograph,charged alesol detectionhttp://ng1.17img.cn/bbsfiles/images/2012/08/201208131731_383588_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208131732_383589_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208131732_383590_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208131732_383591_2352694_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208131732_383592_2352694_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP