请教一个技术问题,我现在做酸碱滴定,用0.1N NaOH滴定醋酸乙酯中的酸度,酚酞指示终点。样品体系为:70%醋酸2%硫酸10%水15%醋酸乙酯其他,杂志据说,使用自动电位滴定仪可以一次滴出醋酸和硫酸的含量,是真的吗?PS,由于体系中有不少的酯,样品制备时不能用水稀释,否则会引起酯的水解。
如题有二甲苯 醋酸丁酯 醋酸戊酯 芳烃 醋酸异丁酯 用什么分离条件好呢?
[em06] 各位大哥大姐:小妹急切寻找以下标准:工业甲醇,活性炭,醋酸乙酯,醋酸丁酯,叔丁胺的国家标准或行业标准.急急急.请大家帮帮忙啦!!!
现在有一个体系,里面有醋酸、水、醋酸乙酯;用FFAP柱子测试,水含量50%~90%,水峰型不好,顶部分叉;醋酸峰型不好,拖尾;用面积归一法定含量,配置样品中醋酸含量50%,测试结果只有34%;求教:这种情况下,是否可以用内标法,用一种内标物同时标定水、醋酸和醋酸乙酯;哪位大神有更好的测试方法?请赐教。
前处理有时加入少量低浓度的醋酸铵的目的是啥阿,另外有个标准当基质为水类时,直接乙腈提取,若为油类时先加正己烷分散,再加70%乙腈提取,是因为乙腈不能溶解油类基质吗,先拿正己烷分散,然后再拿乙腈从正己烷中萃取出目标物质?
醋酸锆溶液中醋酸的测定,用氢氧化钠标准溶液滴定时,酚酞为指示剂,溶液中出现沉淀,计算结果不能肯定。有无其它方法测定醋酸锆溶液中的醋酸,请大家帮忙?提供方法可靠的,本人送软件《化学分析计算软件》的注册码。
我们有一调味姜产品,平日检测总酸(乳酸计),用氢氧化钠滴定。现在要求检测其醋酸含量,我有点疑惑,是同样检测只是把乳酸系数换成醋酸系数吗?就是检测酸度,但是结果计算的时候用醋酸系数计算。请知道的朋友解答一下,先谢了。
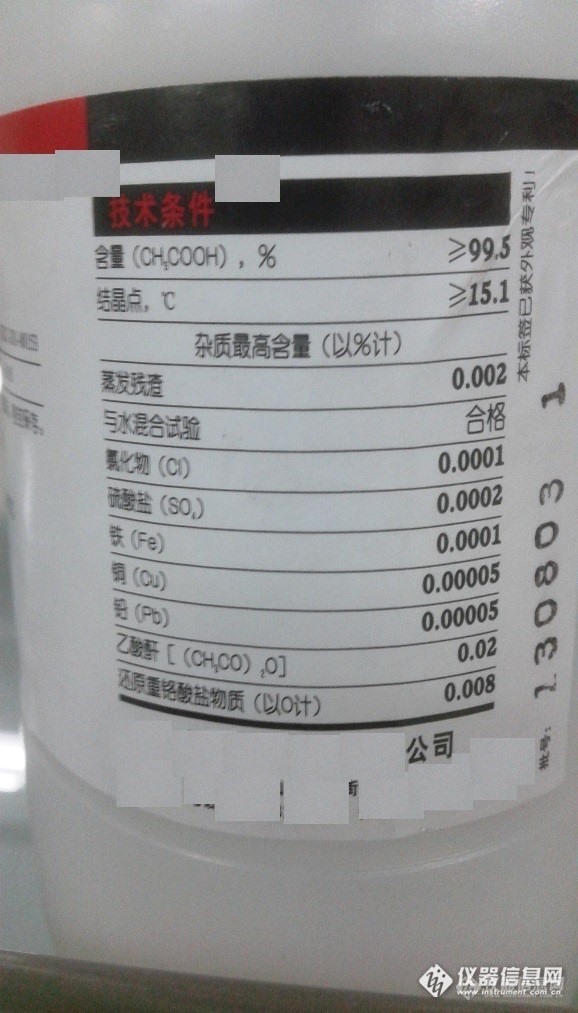
Merck EMSURE用户体验报告系列之一作者:山东某制药公司分析中心 在我公司某制剂有关物质和含量的检测过程中,使用到了醋酸铵(CH3COONH4)和冰醋酸(CH3COOH)两种化学试剂。其中,醋酸铵是有关物质和含量的流动相缓冲盐,冰醋酸是流动相的pH调节剂。这两种化学试剂的质量好坏,对检测结果具有较大的影响。 在试验中,作者发现,采用国内某品牌的醋酸铵和冰醋酸配制缓冲盐进行检测时,色谱图基线噪音较大,使检测方法的灵敏度明显降低。而采用Merck公司的EMSURE系列的优级纯醋酸铵和优级纯冰醋酸配制缓冲盐进行试验,基线噪音明显较小。见图1。http://ng1.17img.cn/bbsfiles/images/2015/02/201502111407_535432_2491887_3.jpg图1 试验色谱图(A.国内某品牌试剂测定图谱;B. Merck EMSURE优级纯测定图谱;C. 国内某品牌试剂测定局部放大图谱;D. Merck EMSURE优级纯测定局部放大图谱;) 试验之余,作者仔细对比了一下Merck EMSURE和国内品牌的醋酸铵和冰醋酸。我惊奇的发现,在这冰醋酸的质量控制指标中,Merck EMSURE优级纯的质控指标竟然多达29项。其中,Merck EMSURE的优级纯冰醋酸共有9种阴离子检测指标,15种金属离子检测指标;而国内某品牌的冰醋酸仅有11项质控指标,其中,4种阴离子检测指标,3种金属离子检测指标。无论是控制的重金属离子种类还是无机阴离子的数量,Merck EMSURE优级纯的质量明显高人一等。从质量控制指标的角度可以看出,Merck EMSURE优级纯的质量具有明显的优势。 空口无凭证,有图有真相。作者将冰醋酸试剂标签上的质控指标拍照展示,大家可以自己进行对比,请看图2。http://ng1.17img.cn/bbsfiles/images/2015/02/201502111408_535433_2491887_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/02/201502111408_535434_2491887_3.jpg图2 Merck EMSURE系列优级纯冰醋酸(左)和国内某品牌分析纯试剂冰醋酸(右) 众所周知,在色谱方法的检测过程中,流动相缓冲盐和pH调节剂等化学试剂的纯度和杂质情况对于检测结果有一定的影响。如果化学试剂中存在一些微量的无机阴离子,则试验所得色谱图的基线噪音会较大,影响检测的灵敏度。缓冲盐中如果存在极微量金属离子如铅、铜、镍、锌、钙、铁、铝等,则会对色谱柱的使用寿命和保留行为产生较大影响。同时,各种杂质的存在往往会对试验结果产生一定的干扰,引起结果的误判。因此,在试验过程中,选择质量更好的优级纯进行化学和色谱分析将会使结果更加准确。
最近做一个四丁基醋酸铵的滴定,在乙酸-乙酸酐介质中用高氯酸进行电位滴定,有很明显的突跃,结晶紫指示剂也变色,但计算结果仅有百分之八十几。样品是我新买的试剂,标注含量有97%。想问问大家看看有没有好的测定方法,或者我的滴定中有哪里出现问题。谢谢。
做酶促反应,得用缓冲液,反应后的体系要进离子阱质谱,工程师说只能用醋酸铵,但试过醋酸铵后反应产物很少,以前用磷酸缓冲液就完全反应了。醋酸铵的缓冲范围是多少?一直用7.4。
请教各位大神:甲苯和醋酸仲丁酯混合后经检测为何只出一个峰值? 甲苯和醋酸仲丁酯单独进样出峰时间都是一样的!后将载气流速调慢,结果是一样的!请问怎样才能将这两种物质区分开来?
请问哪位大虾有没有醋酸钠和醋酸的分析方法?由于是测定废水中的含量,所以用氢氧化钠滴定的方法误差会比较大。
听说用顺铂可以代替醋酸铀对超薄切片进行染色,没有辐射,而且效果也不差,有人知道染色的方法吗?
[size=4]今天同事买了2瓶冰醋酸(分析纯),以前新买的时候都是液体的,这次由于同事的疏忽大意,没有查看,买回来后发现是固体的,不知道大家有没有出现这些情况?究竟新购买的冰醋酸有没有是固体的?你们觉得这能不能退货呢?[/size]
用顶空气相色谱测试烟包VOC含量要用到三醋酸甘油酯,不知道大家都用多少纯度的,在哪里有卖的呢?另外,国标上面的分流比是10:1会不会太大了,5级标样峰会不会比较小呢?有知道的帮下忙啦,新进的仪器,都不太会弄。
[b]‘有奖问答’选择题:在醋酸溶液中加入醋酸钠时,H[sup]+[/sup]离子浓度将会( )。 (A)增大[sub] [/sub](B)不变 (C)减小 (D)无法确定 [/b]
如果要配pH=5, 5mM 浓度的醋酸铵-醋酸缓冲溶液, 我先配好5mM的醋酸铵后用 浓醋酸(99.7%)的醋酸配 pH 配成我需要的就可以吗? 不知道用什么浓度的醋酸来配缓冲溶液是正确的,发现,我把浓醋酸稀释了,加入的量也是差不多的,,可能两种配发配的缓冲溶液的缓冲范围不一样吧。。。 这是要用来做高效液相的流动相的,相信大家也经常配这样的缓冲溶液。。 请帮忙,指点一下,, 怎么配是正确的。。。 醋酸铵-醋酸体系的缓冲溶液的PH范围是从多少到多少呢。。???谢谢,, 急做实验,,,等待。。。。
求醋酸钴的EDTA滴定方法或者有其他更好的方法???用紫尿酸铵终点不明显
经常看到有文章说我们平常吃的醋,虽然吃起来是酸的,但是是碱性食品,真的吗?跟实验室的冰醋酸在分子式和酸碱性有啥区别啊?
做农残检测前处理时候,提取溶剂是入醋酸乙腈和纯乙腈有啥区别啊?另外还想知道提取溶剂是甲酸乙腈或醋酸乙腈区别大吗?分别加入乙酸钠和无水硫酸镁作用一样吗,主要是脱水吗?谢谢大家
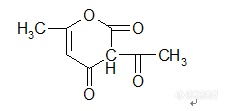
【中文名称】脱氢醋酸;α,γ-二乙酰基乙酰乙酸【英文名称】dehydroaceticacid【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/02/201202191926_349889_1855403_3.jpg【熔点(℃)】108~110【沸点(℃)】270【性状】 白色或淡黄色结晶粉末。无臭,无味。【溶解情况】 难溶于水,在碱性水溶液中溶解度大(20℃时30%以上)。易溶于苯、乙醚、丙酮及热酒精。【用途】 是一种低毒、高效的广谱抗菌剂,在酸、碱条件下均有一定的抗菌作用。可用作食品和饲料的防腐剂。【制备或来源】 由乙酰乙酸乙酯经脱醇缩合而成,或由双乙烯酮缩合制得。【其他】 对光、热稳定。【生产单位】 江苏南通醋酸化工厂;江苏常熟防腐剂厂等
如何将样品中醋酸和醋酸酐的峰分开
在液相反应平衡常数测定中 盐酸 醋酸 醋酸钠 起什么作用
关于非水滴定问题现在一个品种,原标准称样量0.2g,加25ml冰醋酸溶解并加5ml醋酸汞试液,用0.1mol/L高氯酸滴定,现在我们想换成电位滴定,原标准滴定我们样品的含量为100.8%(偏高),现在筛选醋酐加入量是出现以下问题:醋酸:醋酐=1:1 含量为100.8% 醋酸:醋酐=1:3时含量为99.6% 醋酸:醋酐=3:1时含量为102%,请问这是什么原因呢 ,个人认为醋酸:醋酐=1:3时滴定结果比较对,但是和原标准方法差别有点大,如果是因为醋酸中水引起的,也不应该加这么多醋酐啊。
含醋酸氯己定卡波姆基质凝胶剂浑浊问题的解决 最近在做一个含有中、西药的凝胶剂,由于西药成分与凝胶基质不能共存,导致加入后即产生浑浊沉淀,但由于西药成分与中药具有协同作用,能显著起到增强疗效的作用,故而还不得不加,于是漫长的工艺尝试过程展开了,好在黄天不负有心人,问题终于解决了,跟大家分享一下! 概念:凝胶剂是指药物与能形成凝胶的辅料制成均一、混悬或乳状液形的稠厚液体或半固体制剂。凝胶剂有油性和水性之分。水性凝胶剂基质一般由水、甘油或丙二醇与卡波姆、纤维素衍生物等构成。水性凝胶剂是近年来发展较快的剂型,因其具有美观、使用舒适、生物利用度高、稳定性好、不良反应少、不污染衣着等优点。卡波姆基质是水性凝胶剂最常用的基质,此基质对酸、碱、醇都有一定的耐受性;能耐受低温贮存和高压湿热灭菌,但不能耐受盐类;有良好的生物相容性,对眼睛和皮肤没有刺激。 仪器:烧杯、玻璃棒、电子天平、电热磁力搅拌器 配方:卡波姆(基质)、三乙醇胺(成胶碱)、甘油(保湿剂)、乙醇、水(溶剂)、吐温-80(增溶剂)、亚硫酸氢钠(抗氧化剂)、乙二胺四乙酸二钠、中药浸膏、醋酸氯己定。 最初制备工艺:取处方量亚硫酸氢钠、乙二胺四乙酸二钠溶解于适量水中,搅拌下加入处方量卡波姆,继续搅拌至溶胀均匀;取处方量的醋酸氯己定搅拌溶解于与乙醇中,加入处方量甘油、搅拌均匀,加入剩余量的水,搅拌均匀,将此溶液加入到卡波姆溶胀物中,搅匀,加入处方量的中药浸膏,加入处方量三乙醇胺,搅拌均匀。 最初的工艺中当醋酸氯己定溶液加入卡波姆中即刻会产生浑浊现象,为解决问题,我们查阅了大量的关于醋酸氯己定的卡波姆凝胶,但文献报道也不尽相同,有的文献的处方工艺加入顺序也是如此但未谈及沉淀问题,有的文献配方加入顺序有所不同,于是我们尝试了其他的加入顺序。 在配方工艺研究中我们按照文献的方法,先将三乙醇胺加入溶胀好的卡波姆基质中形成凝胶,然后再加入醋酸氯己定溶液,令人头疼的是,沉淀又产生了。没办法,接着尝试,我们将加入顺序重新组合…毫不夸张的说,我们已经将所有可能的加入顺序都尝试了,结果仍无济于事。 经一些专业论坛查询,这种情况不只发生在我们头上,挺多人都遇到了这种麻烦,但并未得到解决。卡波姆为交联聚丙烯酸树脂,显酸性,醋酸氯己定为胍类消毒剂,为碱性,两者混合后会发生反应。看来只有另辟蹊径了。 恰好实验室有人做挥发油的包合试验,因为挥发油气味比较刺激,影响口服效果,所以将其包合,掩盖不良气味。于是我们突发奇想,为什么不将醋酸氯己定包合上再加入卡波姆基质中呢,这样就可以避免两者的直接总接触了。 包合我们采用的是倍他环糊精,考虑到醋酸氯己定的分子量较大,包合比较困难,我们加大了环糊精的比例,摩尔比例为10:1,条件为40℃水浴加热2个小时。经包合后的醋酸氯定溶液再加入到卡波姆基质中,结果真的变好了。功夫不负有心人,问题最终得到了解决。 只是把这次试验的经历大致叙述了出来,描述有些拖沓,请大家见谅,试验总会有问题出现,只要大家不气馁,多思考,多尝试,总会找到解决的办法,可能试验对大家不会有什么帮助,但希望解决问题的思路会对大家有些启发。
请问如何用液质测醋酸氯己定和替硝唑?
二丁基二月桂酸锡和二丁基二醋酸锡等有机锡类能否用某种特定的色谱柱检测?
为什么NaOH标准溶液能直接滴定醋酸,而不能直接滴定硼酸?
有谁知道二乙二醇丁醚和二乙二醇丁醚醋酸酯的极性大小啊,我根据它们的结构,初步判断它们是极性物质,但我用极性色谱柱分离检测时,却发现它们的检测限远远大于它们在非极性柱上的检测限,而且在非极性柱上出的峰形对称又尖锐。有谁能够帮我解释一下啊,谢谢了!
群友提问: 大家好,谁测过醋酸残留啊,直接进样好还是顶空好一些?醋酸响应好低有人回复: 有些样品不方便直接进样像水或者比较脏的样品 那就顶空方便的多。跟大分子量有机酸比响应肯定低 首选WAX柱子群友提问2: 还有个问题,醋酸打进去,貌似在柱子里面不能完全吹干净,对后面进样有影响?有什么解决办法吗?有人回复:那肯定有,你把柱温箱的温度提高 或者做个程序升温不就得了







 我要推广仪器
我要推广仪器
 下载APP
下载APP