我们想用GC来检测乙酰氧基乙酰氯,因为原来是采用滴定方式,过程比较烦琐,而且结果准确度不高。
乙酰二茂铁的合成目的原理实验目的 1 通过乙酰二茂铁的制备,了解用Friendel-Crafts酰基化反应制备非苯芳酮的原理和方法。2 学习柱色谱分离提纯产品和薄层色谱跟踪反应进程的原理和操作方法。实验原理 二茂铁又名双环戊二烯基铁,是由2个环戊二烯负离子和一个二价铁离子键合而成。一般认为,以乙酸酐为酰化剂,三氟化硼,氢氟酸,磷酸为催化剂,主要生成一元取代物;如用无水三氯化铝为催化剂,酰氯或酸酐为酰化剂,当酰化剂与二茂铁的摩尔比为2∶1时,反应产物以1,1′-二元取代物为主。二茂铁及其衍生物的分离最好是用层析法。本实验用柱色谱分离提纯产品,可用薄层色谱法跟踪反应进程,柱色谱和薄层色谱均属于吸附色谱,柱色谱分离提纯是根据二茂铁,乙酰二茂铁和1,1′-二乙酰基二茂铁对活性氧化铝吸附能力的差异而进行分离提纯。用薄层色谱跟踪反应进程,根据二茂铁和乙酰二茂铁的斑点大小可以了解乙酰化反应的进程。仪器药品 5ml圆底烧瓶,克莱森接头,干燥管,电磁加热搅拌器,30cm色谱柱(自制),30×100mm载玻片,离心试管50ml烧杯,玻璃钉漏斗,吸滤瓶,锥形瓶,氮气袋,250ml烧杯二茂铁,乙酸酐,85%H3PO4,25%NaOH,二氯甲烷,棉花,洗净的砂,Ⅲ级活性氧化铝,己烷,醇,硅胶,0.5%羚甲基纤维素,干燥氮气。过程步骤 一、乙酰二茂铁的制备称取100mg(0.54mmol)二茂铁,放入5ml圆底烧瓶中,加入2.0ml醋酸酐。装上带有干燥管的克莱森接头。水浴温热并搅拌使二茂铁溶解。移去水浴,打开塞子迅速加入3ml 85% H3PO4,使反应液变成深红色,室温下搅拌1.5h,在反应期间定期用毛细管在液面上吸取2滴左右反应液放入具塞小试管中,假如10滴二氯甲烷,所得溶液用薄层色谱法展开,以了解反应进程。当二茂铁的斑点很浅时,表示反应基本完成。将反应液滴入盛有1g碎冰5ml烧杯中,滴加25%NaOH中和恰至碱性,得到大量桔黄色沉淀。充分冷却后抽滤,1ml冷水分几次洗涤沉淀,抽干,干燥后称重约110~120mg。二、乙酰基二茂铁的柱色谱法分离(1)干法装柱将粗产品溶于0.5ml二氯甲烷加入300mgⅢ级活性氧化铝,振荡均匀得浆状物。在通风橱中,在干燥氮气下除去溶剂至恒重,得到松散的颗粒状物,精确称取1/2用作柱色谱分离。将自制的1.5×30cm色谱柱洗净,干燥,柱底铺一层玻璃棉或脱脂棉,再铺一层约5~8mm厚的砂,填平。称取5gⅢ级活性的中性氧化铝(60~80目),通过漏斗将氧化铝装入柱管内,轻敲柱管,使之填均匀。将精确称得含有1/2产品重的氧化铝装入柱内,顶部盖一层约5mm厚的砂子,使氧化铝顶端和砂子上层保持水平。(2)洗脱用己烷作洗脱剂从柱顶加入,缓慢滴入己烷逐渐展开得到黄色、橙色分离的色谱带。黄色的二茂铁带首先从柱下流出,用己称重的锥形瓶收集洗脱溶液。当黄色谱带完全洗脱下来时,改用体积比为1∶1的二氯甲烷己烷混合物洗脱,同时橙色带往下移动,逐渐改变溶剂的比例到体积比9∶1二氯甲烷己烷混合溶剂时,则将橙色色谱带完全洗脱下来,用另一只已称重的锥形瓶收集洗脱液。最后改用体积比为9∶1二氯甲烷甲醇洗脱时,可以看到很淡的,很少量的,棕色色带向下移动,将该洗脱液另行收集。(3)收集产品在通风橱内,各组分洗脱液分别在水浴上蒸馏,回收溶剂。浓缩后的溶液放置冷却析出结晶,将产品放在盛有石蜡片的干燥器内至恒重。可回收到未反应的二茂铁20~22mg;得到乙酰二茂铁80~90mg 1,1′-二乙酰基二茂铁少于2mg。分别测定熔点。注意事项1.二茂铁需经升华或用石油醚(30~60℃)重结晶纯化。2.仪器应是充分干燥的。3.乙酸酐是临用前经重新蒸馏的。4.吸附剂的活性与其含水量的关,含水量越低,活性越高。氧化铝放入高温炉中(300~400℃)烘3h得无水物即Ⅰ级氧化铝。Ⅲ级氧化铝可用Ⅰ级活性氧化铝加入重量的6%的水而得到。如所用氧化铝活性过强会使产品不易洗脱,浪费较多的溶剂。5.这里是考虑到柱色谱的容器。一般粗产品重75mg以上都仅取1/2作柱色谱分离。6.二茂铁易升华,故测熔点时要封管。熔点的文献值:二茂铁为173℃,乙酰二茂铁为85℃,1,1ˊ-乙酰基二茂铁为130℃。分析思考1. 二茂铁乙酰化反应的机理怎样?2. 怎样利用薄层层析判断乙酰化反应的进程?3. 乙酰二茂铁在石油醚和乙醚中溶解度哪个更大?为什么?4. 柱层析分离二茂铁衍生物时,如何选择展开的溶剂? [img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52002_1632583_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705162025_52003_1632583_3.gif[/img]
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
求助:请问谁知道乙酰氨基丙二酸二乙酯(医药中间体)测定方法,液相或者[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],或者其他的方法也行,急需,谢谢,我的邮箱是yangxy12345@163.com
请问大家在做卤素测试时,标准EN14582提及到的“白明胶或乙酰丁酸胶囊”和“三氧化二铝”是在哪里买到的?谢谢!
由于手上没有2,3,4-三甲氧基乙酰苯(2,3,4,-trimethoxyacetophenone,C11H14O4)的对照品,所以想求助大家有没有它的光谱图,有的请发一份上来好吗?谢谢大家帮忙啦,
如果要测二氯乙酰氯残留的话是不是可以直接和正丁醇酯化,然后不中和而是直接顶空进样呢,这样反应生成的盐酸也不会进到色谱柱里吧求大佬们指导一下方法可行性,感谢啊!
欧洲食品安全局(EFSA)于近日公布一份科学意见,并在5月16日发布的报道中称:“调味物质3-乙酰基-2,5-二甲基噻吩(3-acetyl-2,5-dimethylthiophene)具有基因毒性(可破坏DNA,即细胞遗传物质),因此关系到人类健康安全。该毒性物质不应该被刻意添加到食物链中。” EFSA并未进行暴露评估,因此其在新闻发布会上表示,消费者从食品中受到的该物质的暴露预计将非常小。 3-乙酰基-2,5-二甲基噻吩被用于给予食品烤坚果味。该物质仅有少数制造商生产,其整体使用率较低(据报告在欧盟内的年使用量为2.3千克)。 英国食品标准局(FSA)已收到通知,虽然英国食品行业该物质的使用量极少,但已对含有3-乙酰基-2,5-二甲基噻吩的食品进行调整。 将该物质从批准调味物质列表中移除的决定受到了所有成员国的支持,并将受到欧洲议会和理事会的监督管制。 欧盟将采纳该决定,并将于7月初生效。此原料在GB2760中,国标编号为S0572 (原编号 I1600),FEMA号为3527.
[color=#444444]请问在分子筛B酸位上形成的甲氧基中的甲基,和乙酰基中的甲基,两者的红外吸收峰有何不同?就是波数怎么变化,哪个高哪个低?非常感谢![/color]
4-氯乙酰乙酸乙酯的检测标准及检测方法N,N-二乙基乙二胺、N,N-二甲基乙二胺、N-乙基乙二胺系列产品,质量按Q/320801GHD001-2004标准执行、含量min.99.%
我公司现急需以下标样,有知道联系厂家的联系方式的请帮帮忙,谢谢!!2,6-二甲酚:纯度大于99.0%3,4-二甲酚:纯度大于99.0%3,5-二甲酚:纯度大于99.0%2,3,5-三甲酚:纯度大于99.0%2,4,5-三甲酚:纯度大于99.0%2,4,6-三甲酚:纯度大于99.0%3,4,5-三甲酚:纯度大于99.0%邻-乙基酚:纯度大于99.0%间-乙基酚:纯度大于99.0%对-乙基酚:纯度大于99.0%萘:纯度大于99.0%联系方式:0531-88032362/88034128联系人:项亮华
[color=#333333]求助。。为什么乙酰乙醛乙酯的质谱中29峰?乙基特征离子峰?[/color]
最近发现固相微萃取提取样品时偶尔出现己二酸二乙基己酯,样品中没有添加,怀疑是之前进样残留,大家有没有遇到过?
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
衷心请教各位高手:1、我们单位即将购进安捷伦GC7890A,配了不同的柱子,为了缩短摸索的时间,想请问用什么类型的柱子做邻苯二甲酸二(2-乙基己基)酯和环氧氯丙烷效果会比较好?2、做这两个项目需要注意些什么?3、国标上做环氧氯丙烷用的检测器是FID,可是据做过的人介绍,好像用FID没办法做出那么低的检测限,不过用ECD或质谱可以做标准物质,但做水样就一直会有干扰峰出现,且跟目标物分不开,请问这干扰物是什么?有什么办法可以分离?衷心期待各位高手能帮我解答,感激不尽!
乙酰二茂铁的液相色谱
本人现急需氯亚铂酸钾和二(乙酰丙酮)铂的分析方法或行业标准,那位大虾有相关资料,请上传或发到我的信箱[color=#DC143C]zhangfy03@126.com[/color],不胜感激!!!!
最近做农残,发现0.5的农残混标中甲胺磷乙酰甲胺磷氧乐果峰小的可怜,我也按照论坛上的方法试了,截取了20厘米的柱子,换了新衬管和进样垫,重新配了1ug/ml的标样,还是那样,这几种物质峰特别小,请大家帮忙分析下原因,急!!!
各位老师,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲胺磷、乙酰甲胺磷、氧乐果出的峰非常低,有时几乎看不见。更换了新称管那些还好一点,进样几次后又几乎不怎么出峰了,有什么办法解决下吗?
上来几批样品,标品中甲胺磷,乙酰甲胺磷,氧乐果就消失不见了,每次维护完仪器(更换衬管,清洗离子源,老化色谱柱)才能恢复,前面以为是仪器有污染了吸附,这一次都还是,就5-60个样品,后面就自动消失了。用的安捷伦7000
帮忙求助乙酰氨基丙二酸二乙酯 的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]与液相测试方法文献
谁有:过氧化苯甲酰和N,N-二羟乙基对甲苯氨的标准红外光谱图。
有誰可提供DMAC(二甲基乙酰氨)之 dew point, 非常感謝!
761做农残,之前做了大半年,柱子氧乐果不出峰了,没办法截了快两米,终于好了。可是发现做了两批样一百多个样,出峰面积又小了。走丙酮溶剂发现有小杂峰(面积很小,50左右。破罐子,破摔。各种折腾。用丙酮洗(进4微升),甲醇洗(进4微升)。老化的时候,进溶剂,甲醇,正己烷,丙酮轮着。结果更惨了,本来氧乐果1微升能出2000多的。现在就八百了。乙酰甲胺磷本来好好的,也少了很多。之前也发现,柱子老化后。本来不拖尾的峰,拖尾了。问各位老师,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱,走溶剂空白,能起到清洗作用吗?还是我用的溶剂体积太大了,能用甲醇吗?以后这种情况要每次做了,截了柱子后,再老化吗?
GC450瓦里安气相,PFPD检测器,DB1701(30*0.25*0.25)柱,进样口温度250℃,检测器300℃;程序升温:80℃保持1min,20℃/min升至130℃,5℃/min升至200℃,15℃/min升至250℃,保持1min,共21.83min。乙酰甲胺磷标液是新打开的,浓度100ug/mL,用丙酮配制成1.0ug/ml进样,乙酰甲胺磷不出峰,但配制的二唪农0.1ug/mL出尖峰。谁做过乙酰甲胺磷,用什么溶剂,它在0.1ug/mL时就出峰?
[b]《空气和废气监测分析方法》(第四版增补版) 环氧氯丙烷 乙酰丙酮分光光度法 [b]环氧氯丙烷的标准曲线哪个大神有?[/b]扩项急用,万分感谢[/b]
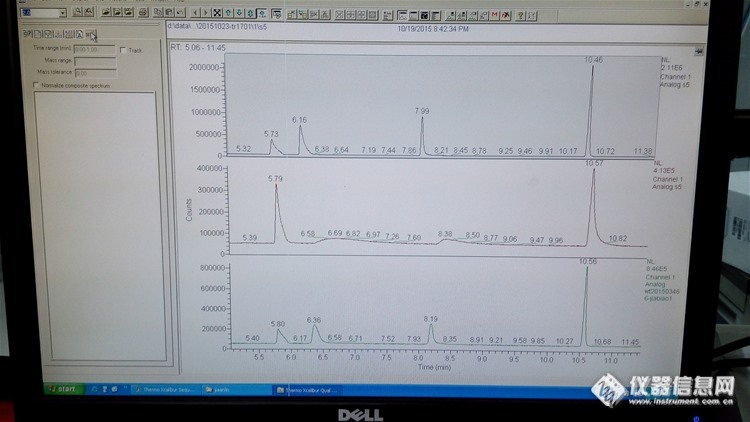
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
GC7890A DB1701新柱以及用了一年后截去1米半之后的柱子,FPD检测器,检测器已清洗 进样口已清洗 衬管全新,分流平板全新。5PPM的乙酰甲胺磷峰高1500左右,0.4PPM的峰高180,再低不出峰了。是1701这根柱子对于乙酰甲胺磷吸附很严重,或者说这根柱子不适合分析此种农药。希望大家交流下,说说你们选择分析有机磷农残的毛细柱和色谱条件!
[color=#444444]遇到一个大大的问题做试验做了7个月了,就是检测不到大米中的2-乙酰基-1-吡咯啉,不出峰,用的spme萃取,热电的质谱,DB wax柱子,以前有老师在日本做的效果十分明显,用的铂金埃尔默的质谱,其他的都一样,当然地点也不一样,请大家帮我分析一下,这是什么原因造成的?谢谢[/color]
在做氯乙酰氯[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]时,我的进样针用完后,用丙酮洗过,第二天再用,进样针拔不出来了,坏了,是什么情况呀







 我要推广仪器
我要推广仪器
 下载APP
下载APP