求助:原乙酸三甲酯分析方法或分析标准
原乙酸三甲酯中微量氯化铵杂质任何去除,因为微量氯化铵的存在影响原乙酸三甲酯的产率,有那位高手指导我,谢谢!
丁酸戊酯丁酸异戊酯异丁酸戊酯异丁酸异戊酯这四个从质谱上 有什么明显的区别吗? 公司没有这些原料 我没法进仪器区分 是刚才分析样品是看到的一个峰,区分不开到底是啥, 大家支支招 还是要我把谱图上传 都可以的。。。。上个附件,RT:12.577 大家有空帮忙看看 这个到底是什么??
如题:七氟丁酸和三氟乙酸能进GC吗?
大家过年好。我最近在做链霉素,用到了七氟丁酸(离子对试剂),我想问一下,七氟丁酸在液质中的作用是什么?使用时需要注意那些事项?谢谢各位能够提供相应的资料。
最近刚开始做[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url],对于很对东西都是边学边做,对衍生化不太熟悉,我用的是BSTFA-TMCS加吡啶70℃下水浴30min,进样1微升,跑了混合的,出了四个峰(不知道17分钟之后那个小的算不算,响应值低于25)我就以为正好,结果单个进样的时候对不上,有没有人做过这个呀,或者有没有大佬说一下可能的原因呢,下面依次是混合,月桂酸,月桂酸单甘脂,三月桂酸甘油酯,三丁酸甘油酯。[img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356375_1876_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033355547_6193_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356248_8395_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033355761_4184_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206171033356826_8013_5654520_3.png[/img]
请问大家在做卤素测试时,标准EN14582提及到的“白明胶或乙酰丁酸胶囊”和“三氧化二铝”是在哪里买到的?谢谢!
45.80646.423是丁酸香茅酯 还是 阿芬美酯?糊涂了 http://simg.instrument.com.cn/bbs/images/brow/em06.gif
关于游离脂肪酸检测,我将游离脂肪酸丁酯化处理,使用硫酸/正丁醇然后发现检测结果丁酸丁酯含量很高,我做了空白以及拿没有丁酸的标品做丁酯化都发现有丁酸丁酯,这丁酸丁酯是怎样产生的啊?有什么方法避免这种情况发生?丁酸是我很重要的目标分析物这样就很难对其定量。(我完成了丁酯化的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]方法开发换方法不可能了)急需大神解答。
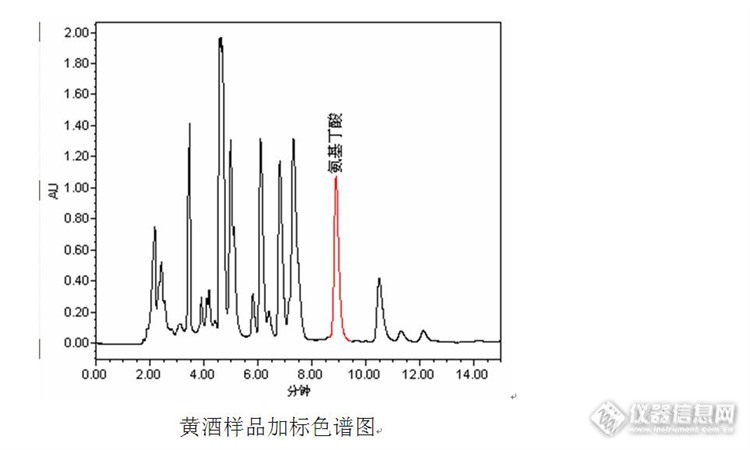
黄酒中γ-氨基丁酸含量测定的辛酸历程 近日实验室收到一批黄酒样品,该批黄酒是用发芽糙米为原料酿造而成,客户要求测定黄酒中的γ-氨基丁酸含量。由于之前实验室以丹磺酰氯为衍生试剂,建立了高效液相色谱法测定发芽糙米中γ-氨基丁酸含量的实验方法,并对实验方法的线性、精密度以及回收率进行了确认,均可以满足发芽糙米中γ-氨基丁酸含量测定要求,因此拿到黄酒样品后直接按照发芽糙米的前处理方法和色谱方法进行分析。链接如下:http://bbs.instrument.com.cn/shtml/20141226/5591256/。然而事与愿违,在测定的液相色谱图中压根就没有见到γ-氨基丁酸的色谱峰,反而在11.5min左右有个小的色谱峰,其峰高与发芽糙米中γ-氨基丁酸峰高有点相似,初步怀疑是保留时间发生了漂移,与发芽糙米样品色谱图对比后发现,在发芽糙米样品色谱图中该保留时间处也出现了一个相似的小峰,因此将该色谱峰是γ-氨基丁酸的可能性排除。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311333_530568_1669358_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412311334_530570_1669358_3.jpg 原本该实验到此结束,准备将实验结果反馈给客户:黄酒中γ-氨基丁酸的检测结果为“未检出”。为了保证数据的准确性和可靠性,在黄酒样品中进行加标实验,结果在加标的色谱图中也未在相应的保留时间出峰,而且11.5min左右的色谱峰也没有增大,因此决定先将“未检出”的结果搁置,并对实验方法进行分析。 经过对样品前处理过程和色谱方法的分析,觉得可能造成加标样品中γ-氨基丁酸未检出的原因可能有:(1)保留时间漂移。由于流动相需要调节pH值,同时样品前处理过程中也涉及到酸、碱溶液的使用,怀疑是流动相或者样品pH的改变导致保留时间的漂移,从而未在原有的保留时间出现应有的色谱峰。然而重新配制流动相和前处理样品,加标样品测定结果依然是“未检出”,对比加标和不加标样品的色谱图,两者几乎一样,也没有峰面积或峰高变化明显的色谱峰;(2)衍生试剂失效。丹磺酰氯对光和湿敏感,不稳定,放置时间久了会生产二氯亚砜并继续分解成其他物质,影响其在有机溶剂中的溶解度,也会影响结果。可是为了排除衍生试剂的问题,重新打开一瓶刚购置不久的丹磺酰氯试剂,并重新试验,结果仍然不理想;(3)衍生条件控制不当。之前用相似的方法测定牛磺酸含量以及测定发芽糙米中γ-氨基丁酸含量时曾出现过衍生过程条件控制不当造成衍生不完全或者不能衍生的情况,可是与黄酒样品同一批处理的γ-氨基丁酸标准溶液和发芽糙米样品均能衍生成功,并正常出峰,为何唯独黄酒样品不出峰呢?在百思不得其解之际,看到同事在滴定黄酒中总酸,忽然间若有所悟:黄酒中的γ-氨基丁酸需要在碱性条件下才能与丹磺酰氯发生衍生反应,而黄酒是酸性介质,pH值一般在3~5之间,同时黄酒为酿造产物,对酸碱性具有一定的缓冲能力。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311336_530572_1669358_3.jpg 通过比较发现:黄酒为酸性样品,缓冲能力较强,按照发芽糙米样品前处理方法直接加入0.5mL 碳酸钠(pH9.8)可能不能达到合适的衍生反应条件,最终导致黄酒样品中γ-氨基丁酸“未检出”。 找到问题后调整实验方案,先将黄酒样品调整至中性,然后再按照发芽糙米样品方法进行前处理。调整实验方案后,黄酒样品中γ-氨基丁酸测定的色谱图如下图。从色谱图中可以发现,经过实验方案的调整黄酒样品中检出了γ-氨基丁酸的存在。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311337_530573_166
做液质,新项目开发,流动相中加了七氟丁酸。现在再进氯霉素(负离子源)不出峰了,怎么办。
我们有一种产品要用到乙酰乙酸甲酯和异丁酰氯,然后在回收乙酰乙酸甲酯时其中会有异丁酸的存在,而这个异丁酸对我们的反应有很大的影响,导致回收的乙酰乙酸甲酯不能使用,请问大家在这种情况下我们可以怎么样除去其中的异丁酸呢?谢谢赐教!
4氯丁酸用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做不出峰,换成[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url],4氯丁酸变成了1,4丁内酯,关键我需要将两者都分开测,有做过4氯丁酸的吗?
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
请教α-酮丁酸溶液的配置经验。各位高手们: 小弟我最近遇到个难题, 配α-酮丁酸溶液的时候发现这药品性状是熔融状态的,称量时用药匙一舀就沾一药匙,甩都甩不下来,而且好中的刺激性气味. 而改用其钠盐做替代物又确实划不来,价格贵了近7倍,且效果没什么太大差别! 哪位有经验的能介绍下好方法. 先谢谢了!![em06] [em06]
吡虫啉的测定与柠檬酸和柠檬酸三甲酯的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911131345_184266_1896702_3.jpg
请问下面的结构如何发音(读音): 3-甲基丁酸读作3(什么?)甲基丁酸
我要分析发酵产物中以下几种产物:丙酮、乙醇、丁醇、乙酸和丁酸。用的是“10% Carbowax-20 M, 0.10% H3PO4,support 80/100Chromosorb WAW” 玻璃填充柱和FID检测器。 因为发酵的底物中有一定浓度的糖类物质,所以采取别人的建议在衬管(liner)中填充了石英棉以阻止糖类物质进入柱子,否则糖类物质会很快的污染衬管而出现奇怪的峰。现在发现另外一个问题:那就是乙酸和丁酸总有残留,也就是说在测完一个样后,如果再测一个水样,会出现乙酸和丁酸的峰,而且峰面积比较大。不知道是乙酸和丁酸残留在柱子中,还是liner中或者其他什么地方?是否因为温度不够高?用的方法:进样口温度 225 oC;检测器温度:225 oC;柱温箱(oven)初始温度 40 oC,ramp 1: 40 oC/min for 3min, 最终温度 200 oC for 7 min; 不知有没有人有类似经历,可以给出您的建议解决这个问题?多谢!
γ-氨基丁酸和α-羟基丙酸氢键核磁峰位置大概是多少?
请教α-酮丁酸溶液的配置经验各位同行: 小弟我最近遇到个难题, 配α-酮丁酸溶液的时候发现这药品性状是熔融状态的,称量时用药匙一舀就沾一药匙,甩都甩不下来,而且好中的刺激性气味. 而改用其钠盐做替代物又确实划不来,价格贵了近7倍,且效果没什么太大差别! 哪位有经验的能介绍下好方法. 先谢谢了!!
请问专家:在南京哪个地方可以检测γ-氨基丁酸呢?
最近公司新进一个原辅料,4-溴丁酸乙酯有没有同行知道该产品相关的GC 检测方法,发给我参考一下,谢谢
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]测氨基丁酸的文献
请问专家:在成都哪个地方可以检测γ-氨基丁酸呢?
有哪位老师咋知道环境空气中的丁酸和二甲基亚砜的测量方法,请求告知
本人最近接手了一个检测药品中4-氯丁酸的分析方法开发的活,单独4-氯丁酸的话没什么,主要是定量限要做到0.5ppm以下,大约5ng/mL的量,所以必须用质谱来做。1mg/mL的浓度负离子模式下虽然有找到目标峰,但是响应并不高。所以只能考虑通过衍生的办法在正离子模式下提高响应,目前使用的衍生试剂为2-巯基吡啶。反应机理大家想必知道(氯上发生取代反应),但是在碱性环境下60℃衍生4h后发现没有衍生上去,加大碱的量升高反应温度均无明显效果。但是我之前做的5-氯戊酸是能衍生上的。我很郁闷差一个碳差距就那么大吗? 希望有经验的前辈多指导我一下,不管是反应机理上的还是有衍生之外的新思路都在这里谢过了!
我们的污水处理,需要用到一种原料:微营养剂。不知道同行的有没有做过微营养剂中正丁酸含量的测定。
最近刚开始[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url],对衍生化不太了解,我用的是BSTFA+TMCS进行衍生化的,混合的出了四个峰(最后那个17分钟之后的不知道算不算峰,响应值<25),我以为正好,结果跑单个的对不上了,下面的图依次是混合,月桂酸,月桂酸单甘脂,三月桂酸甘油酯,三丁酸甘油酯。有没有做过的呀[img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905473939_5594_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475042_7945_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475286_2664_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475296_4906_5654520_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/06/202206170905475101_6177_5654520_3.png[/img]
气相测定三甲胺时,三氯乙酸起什么作用呢?急
如何让丁酸、戊酸、异戊酸分开?







 我要推广仪器
我要推广仪器
 下载APP
下载APP