环氧溴丙烷内标物(DMF为溶剂),反应时间为3小时,温度为80摄氏度。
衷心请教各位高手:1、我们单位即将购进安捷伦GC7890A,配了不同的柱子,为了缩短摸索的时间,想请问用什么类型的柱子做邻苯二甲酸二(2-乙基己基)酯和环氧氯丙烷效果会比较好?2、做这两个项目需要注意些什么?3、国标上做环氧氯丙烷用的检测器是FID,可是据做过的人介绍,好像用FID没办法做出那么低的检测限,不过用ECD或质谱可以做标准物质,但做水样就一直会有干扰峰出现,且跟目标物分不开,请问这干扰物是什么?有什么办法可以分离?衷心期待各位高手能帮我解答,感激不尽!
作液化石油气(主要组分是丙烷)分析,用三氧化二铝大口径毛细管柱分析。但是工艺要求检测苯,含量大约在5ppm左右,请问那位大侠知道苯能否用此柱分析?如果能操作条件是?谢谢
请问:气相色谱法检测苯和环氧氯丙烷残留溶剂的条件该怎样设置? 若采用顶空进样,程序升温方式,该选择什么样的柱子,以及程序升温方式怎么设定?
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
[color=#444444]大侠求助,怎么用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的方法对溴甲基环丙烷进行含量测定,急急急。[/color][color=#444444] 请给出载气、流速、柱温等具体测定条件哦。。。[/color][color=#444444] 谢谢啊[/color]
请问有没有在GC上做过这样的体系:四氢呋喃和溴丙烷的混合物?两者沸点很接近,四氢呋喃64.5度,溴丙烷71度。求教各位。谢谢!
最近老板让做消毒副产物卤乙酸,同事订了瓶1,2-二溴丙烷作内标用的,这瓶1,2-二溴丙烷瓶子上就标识了97%,100g。按实验要求是要配成300ug/L的内标萃取液,麻烦各位老师教教我怎么配啊? PS:我看到有人说取0.3g,然后定容什么的,现在是我不知道这个买回来的溶剂的体积是多少,没法具体定量啊。。实在是第一次接触这些东西,还请各位大侠指导我一下。谢谢 [img]http://simg.instrument.com.cn/bbs/images/brow/em09509.gif[/img]
环氧氯丙烷最早于1854年由Berthelot用盐酸处理粒甘油,然后用碱液水解时首先发现的。20世纪60年代前后,为适应环氧树脂生产发展的需求,环氧氯丙烷开始以氯丙烯为原料作为主要产品进行生产。工业上环氧氯丙烷的生产方法主要有丙烯高温氯化法和醋酸丙烯酯法两种。前者由美国Shell公司于1948年首次开发成功并应用于工业化生产,当前世界上90%以上的环氧氯丙烷采用该方法进行生产。后者由前苏联科学院以及日本昭和电工公司于20世纪80年代分别开发成功。 生物柴油衍生副产甘油的大量产出,甘油路线生产环氧氯丙烷工艺成本上得以可行。该路线突出的优势是绿色环保,三废量较丙烯法得到极大减少,索尔维、江苏扬农、益海嘉里等企业是该工艺路线的领军者。 危险性摄取,吸入及皮肤吸收有毒。刺激性强烈。可能会致癌。在空气中容许量2ppm。易燃,中度着火危险性
请教各位:谁知道溴代环丙烷的色谱分析方法,谢谢!
各位前辈,我现在要开展环氧氯丙烷和氯苯,我们单位有安捷伦的DB-5、DB-1701、DB-WAX三种色谱柱,诚恳请教要测这两种物质应该选择什么色谱柱?如果这三种色谱柱不合适,建议买哪种色谱柱?谢谢!!!!
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]测卤代烃,用乙酸乙酯作溶剂,为什么内标物-溴丙烷出不来,用的是DB-5MS的毛细管柱,内标物的浓度为1000ug/mL都出不来,溶剂峰很大。是溶剂峰太大了,把内标覆盖了?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/03/202303151117246002_3229_5195900_3.png[/img]
我现在要检测环氧氯丙烷的水份,但不想用GB的GC法(没有TCD检测器),现用KF法检测时,甲醇会与样品反应造成检测结果偏高。我想请老师们指点一下,用KF做环氧氯丙烷的水份,最好用哪种溶剂好??????????
谁有1.3-溴氯丙烷(cas:109-70-6)的氢谱图
求助,我想知道HP-5或者SE-54的毛细管柱(两种色谱柱的填充相都是5%-二苯基-95%-二甲基硅氧烷毛细管柱),能不能将甲烷、乙烷、乙烯、丙烷和丙烯分开?
[font=SimSun, STSong, &]降甜剂即2-(4-甲氧基苯氧基)-丙酸钠,这种香料的实际作用只是使别人口感上觉得甜度下降了还是说它跟糖有了化学反应造成了糖的转化?[/font]
丙酮中环氧氯丙烷标准物质,坛墨的,100微克每毫升,色谱图怎么是这样的,哪个峰是环氧氯丙烷,求大神帮忙指点一二,跪谢!!![img]http://ng1.17img.cn/bbsfiles/images/2018/08/201808101748188893_6544_3237457_3.jpeg[/img]
GB5749-2006检测水中环氧氯丙烷,样品前处理采用二氯甲烷。我实验后过效果很差。参照EPA方法,采用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]可直接进样,我用的是乙腈中的环氧氯丙烷,可是用水稀释后就找不到色谱峰了。请教各位,急待解决!
[color=#333333]环氧氯丙烷是用二氯甲烷做溶剂的,但是我没有,可以用二硫化碳代替吗?[/color]
刚做气相,做环氧氯丙烷的方法,结果不出峰,请教各位专家。情况如下安捷伦7890A,顶空进样,FID检测器顶空条件:传输温度142 ℃,炉温60℃,加热时间10min色谱条件:进样口150℃;柱温,初始40℃,保持4min,以30℃/min升至150℃,保持2min;检测器FID ,250℃。柱:安捷伦19091J-413 (HP-5)标准溶液:环氧氯丙烷用二氯甲烷配制,浓度497mg/L;取标液50微升加入盛有10.00mL纯水的顶空瓶中,密封,进样。结果发现只有溶剂峰二氯甲烷的峰,没有别的峰,怎么回事?
我在做一产品,此产品中含大量的环氧氯丙烷溶剂。现在单进环氧氯丙烷样品时,乙腈/水的流动相,UV210时竟出现两个峰。一个为4分钟,另一个为9.5分钟。应该是波长不对,可是没办法进行扫描了,不知哪个峰是环氧氯丙烷的组份。请老师帮忙,有什么好办法,进行确定啊。

在做原料中环氧乙烷和环氧丙烷的检验方法,遇到了限度问题,想请教一下,一方面,在2020版药典四部,所有对环氧乙烷进行检验的方法里限度都是0.0001%(1ppm)另一方面,在ICH M7(R1)中的第26页,恰好以环氧乙烷为例演示了限度值是怎么计算的,就是以CPDB查询到的TD50来计算限度,最后计算结果是21.3微克/人/天,如下面的图,这样不就是对应原料限度是21.3ppm吗?所以,我应该以1ppm还是21.3ppm为限度呢?环氧丙烷按照ICH的方法,根据TD50计算是74.4ppm,药典四部有两个品种做环氧丙烷,限度分别是0.0005%和0.001%,环氧丙烷做的比较少。但是都比按TD50计算的结果小很多。欧洲药典中曲克芦丁有检环氧乙烷,限度也是1ppm,所以这个1ppm是出自哪里呢?是否应该按1ppm来作为限度呢?有帖子说05版药典残留溶剂附录中对环氧乙烷有限度规定,但现在的2020版或2015版中都没有查到,是否因为环氧乙烷是基因毒性杂质,所以没有在残留溶剂里面见到了,而ICH M7是对基因毒性杂质进行说明的。所以究竟以1ppm还是以21.3ppm做限度呢?[img=,650,899]https://ng1.17img.cn/bbsfiles/images/2021/03/202103081021496056_5338_2789522_3.jpg!w650x899.jpg[/img]
[color=#444444]各位大神,跪求环氧氯丙烷可以测紫外图么?如果可以,它会在哪里出峰,跪求一张环氧氯丙烷的紫外吸收光谱图。另外,还需要一张黄腐酸的紫外吸收光谱图,谢谢了![/color]
由于是六通阀进样,实验有需要测定环氧丙烷的校正因子,由于标气太贵了,想自己来测定。在文献中有看到将环氧丙烷放在冰水浴的饱和器(saturator)中,借助氮气吹到六通阀内进行单点定量。这样的方法可行吗?我想问的是:1. 饱和器是什么,我想是不是就是一个密闭的瓶子2. 氮气的流量会不会影响饱和蒸气的浓度?我想是氮气的流速要低于环氧丙烷的挥发速度才能保证出来的气体是环氧丙烷的饱和蒸汽,若有经验的话望能得到解答。
请教大家如何配置和稀释气体,样品是环氧乙烷、环氧丙烷、环氧氯丙烷
谁有环氧丙烷的分析方法啊?有知道的吗?可以告诉我吗?谢谢啊!
水中环氧氯丙烷、氯乙烯、甲醛、乙醛、丙烯醛、二硝基苯、硝基氯苯和2,4,6-三硝基甲苯的监测方法([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]),包括仪器、试剂。
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]吹扫捕集做环氧氯丙烷不出峰
[b]《空气和废气监测分析方法》(第四版增补版) 环氧氯丙烷 乙酰丙酮分光光度法 [b]环氧氯丙烷的标准曲线哪个大神有?[/b]扩项急用,万分感谢[/b]
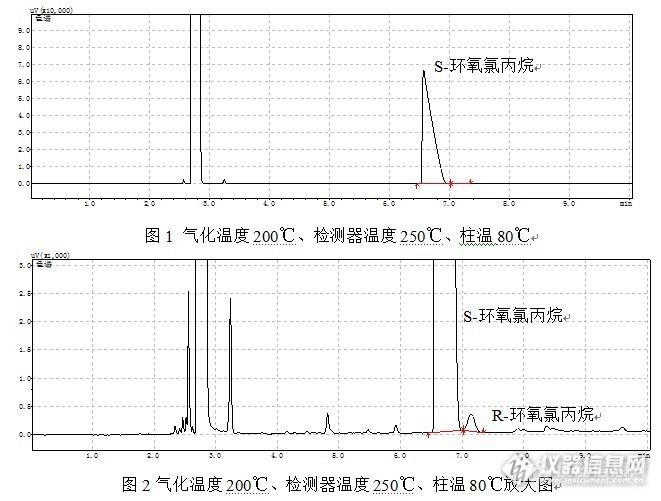
请教各位战友,现用手性气相色谱柱分析S-环氧氯丙烷的手性纯度,色谱条件:进样口200度,柱温80度,检测器250度,氮气流速1.4 ml/min,分流比30:1。S-环氧氯丙烷与R-环氧氯丙烷分离良好,但是S-环氧氯丙烷峰形特别难看,拖尾因子有5。升高柱温至100度,S-环氧氯丙烷拖尾因子也有2,而且S,R构型分不开。请教,1.色谱条件还能如何优化,才能在峰形与分离度之间达到平衡?2.总觉得S-环氧氯丙烷峰形特别奇怪,直线上去,斜线下来,这样正常吗?是色谱系统有问题吗,还是其他原因导致?谢谢!http://ng1.17img.cn/bbsfiles/images/2013/11/201311221340_478814_1605035_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP