
盐酸可乐定的测定和盐酸丁咯地尔片含量的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021833_180179_1896702_3.jpg
各位老师,求帮忙,,,我用高效液相色谱分析盐酸地尔硫卓,发现峰型拖尾严重(拖尾因子都有4.9)流动相:醋酸盐缓冲液(用氢氧化钠调PH6.2):乙腈:甲醇=50::25:25用无水乙醇溶解的样品,浓度为0.1㎎/ml柱子是新的C18,250X4.6的
硫酸-盐酸底液极谱法测锑时,标准和样品起波的位置不一样是什么原因
我用盐酸萘乙二胺法做亚硝酸盐检测时,发现当我加入盐酸萘乙二胺后,出现红色显色反应,但是过30s左右,颜色随即消失,这好象不符和一般的显色时间的原理,请问这可能是什么原因,谢谢!
GB/T6432-94标准中4.6 盐酸标准溶液:邻苯二甲酸氢钾法标定,按GB601制备。平常都是用碳酸钠标定盐酸,用邻苯二甲酸氢钾标定氢氧化钠,这里却.......该如何理解呢?
名 称 盐酸滴定液的配制及标定操作规程 一、 目 的:建立盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、 0.1mol/L)的配制及标定的标准操作规程,确保检验的准确性。二、 适用范围:适用于盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、0.1mol/L)的配制及标定。三、 责 任 者:QC化验员、QA。四、 引用标准:中华人民共和国药典(2000年版)二部附录。五、 规 程:1、误差要求: 法定标准标定 标定份数≥3份,相对偏差≤0.1%复标 复标份数≥3份,相对偏差≤0.1%标定、复标 二者的相对偏差≤0.15%滴定液浓度 (F)应在1.000-1.050之间2、反应原理:2HCl+Na2CO3 2 NaCl+H2O+CO23、指示剂:甲基红-溴甲酚绿混合指示液4、基准试剂:基准无水碳酸钠5、仪器与用具:三角烧瓶、滴定管6、操作步骤配制:盐酸滴定液(1 mol/L) 取盐酸90 ml,加水适量使成1000 ml,摇匀。 盐酸滴定液(0.5 mol/L、0.2 mol/L或0.1 mol/L) 照上法配制,但盐酸的取用量分别为45 ml、18 ml或9.0 ml。标定:盐酸滴定液(1 mol/L) 取在270~300 ℃干燥至恒重的基准无水碳酸钠约1.5g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示液10滴,用本液滴定至溶液由绿色转变为紫红色时,煮沸2分种,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml盐酸滴定液(1mol/L)相当于53.00mg的无水碳酸钠。根据本液的消耗量与无水碳酸钠的取用量,算出本液的浓度,即得。盐酸滴定液(0.5mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.8g。每1ml盐酸滴定液(0.5mol/L)相当于26.50mg的无水碳酸钠。盐酸滴定液(0.2mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.3g。每1ml盐酸滴定液(0.2mol/L)相当于10.60mg的无水碳酸钠。盐酸滴定液(0.1mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.15g。每1ml盐酸滴定液(0.1mol/L)相当于5.30mg的无水碳酸钠。如需用盐酸滴定液(0.05 mol/L、0.02 mol/L或0.01 mol/L)时,可取盐酸滴定液(1mol/L或0.1mol/L)加水稀释制成。必要时标定浓度。7、计算公式: 盐酸滴定液(1 mol/L) F=M/(V×0.053)盐酸滴定液(0.5 mol/L) F=M/(V×0.0265)盐酸滴定液(0.02mol/L) F=M/(V×0.0106)盐酸滴定液(0.1 mol/L)F=M/(V×0.0053)式中:M无水碳酸钠的质量(g) V滴定所耗盐酸滴定液的体积(ml) 8、注意事项反应致近终点时,应加热煮沸2分钟,以赶走溶解于水中的CO2,排除CO2对反应的干扰。
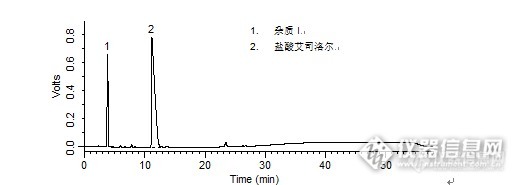
1. 杂质I2. 盐酸艾司洛尔 盐酸艾司洛尔样品制备 制备方法有关物质衍生溶液:取盐酸艾司洛尔对照品约10 mg,置10 mL量瓶中,加入1 mol/L盐酸溶液1 mL,放置30分钟,加1 mol/L的氢氧化钠溶液1 mL使中和,用流动相A 稀释至刻度,摇匀。分析条件 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相流动相A:乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0 g,加水至650 mL)=15:20:65流动相B:甲醇梯度流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 色谱图有关物质衍生溶液http://ng1.17img.cn/bbsfiles/images/2016/04/201604211737_591070_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 3.842 6280189 655879 2747.059 0.670 -- 2 11.157 29271705 784686 1512.532 5.026 10.154 本品种同时使用了SpursilC18色谱柱,在药典规定条件下进行检测,满足药典要求。
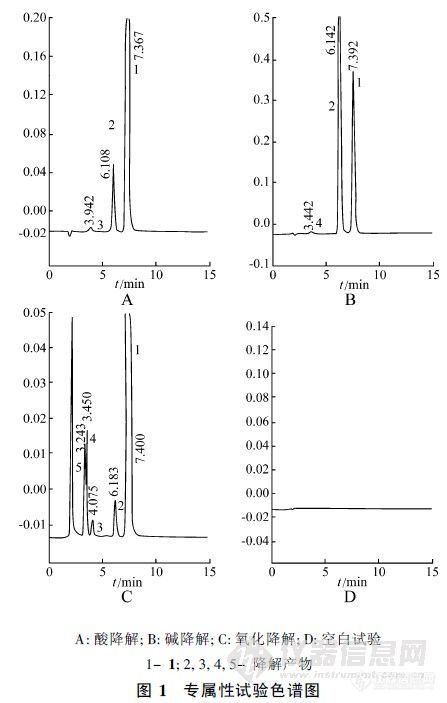
【作者】 王璐璐; 郑稳生; 谭小川; 张宇佳;【Author】 WANG Lu-Lu, ZHENG Wen-Sheng, TAN Xiao-Chuan, ZHANG Yu-Jia (Chinese Academy of Medical Sciences, Institute of Materia Medica, Beijing 100050)【机构】 中国医学科学院协和医科大学药物研究所; 中国医学科学院协和医科大学药物研究所 北京100050; 北京100050; 北京100050;【摘要】 采用 HPL C法测定注射用盐酸地尔硫的含量及有关物质。色谱柱为 Diamonsil C1 8( 15 0 mm× 4.6 m m,5μm ) ,0 .1m ol/ L醋酸钠缓冲液 ( p H6 .2 ) -甲醇 -乙腈 ( 5∶ 6∶ 6 )为流动相 ,检测波长为 2 40 nm。检测限为 0 .2 ng,线性范围为 6~ 14μg/ ml( r=0 .9998) ,RSD为 0 .11% ,平均回收率为 10 0 .2 % ,RSD为 0 .5 2 %。 更多还原【Abstract】 A HPLC method for the determination of diltiazem hydrochloride for injection and its related substances was presented. Diamonsil C18 column was used, with mobile phase of 0.1 mol/L sodium acetate buffer solution (pH6.2)-methanol-acetonitrile (5∶6∶6) and at detection wavelength of 236 nm. The detection limit was 0.2 ng. The linear range was 6~14 μg/ml, and average recovery was 100.2% with RSD of 0.52%. 更多还原【关键词】 注射用盐酸地尔硫; 有关物质; HPLC; 测定; 【Key words】 diltiazem hydrochloride for injection; related substance; HPLC; determination; http://ng1.17img.cn/bbsfiles/images/2012/08/201208201140_384595_2352694_3.jpg
HPLC法测盐酸普萘洛尔含量,采用紫外检测器,用安捷伦-SB-C18柱,以乙腈:0.05M磷酸二氢钾30:70为流动相,但出来的色谱峰拖尾,且峰形对称性差。本人试过加酸加碱来改善拖尾,但效果不明显,特此求助,望有关人士能解我燃眉之急,非常感谢!!

http://ng1.17img.cn/bbsfiles/images/2014/01/201401052234_486748_2555187_3.jpg以上为GB/T 601中对滴定溶液结果处理时的要求。最近一次外审,审核老师要求盐酸滴定溶液数据严格按照GB/T 601要求来处理,于是来论坛寻求处理方法,发现有两种做法(产生分歧的原因是对标准中文字表述的理解不同):以四平行为例,“每人四平行测定结果极差的相对值,不得大于重复性临界极差的相对值0.15%"1.重复性临界极差的相对值等于0.15%,直接计算极差的相对值X,比较X与0.15%大小;2.分别计算极差的相对值X和重复性临界极差的相对值Y,比较X与Y乘以0.15%的大小。我按照第一种做法算了下: 极差相对值等于四次结果极差除以四次结果平均值。假设四次结果平均值为0.07,则四次结果极差不得大于0.07乘以0.15%,即0.000105。这个数字太小,小到即使你滴定只有半滴之差也满足不了!第二种做法,论坛有现成的模版,请见附件。不知道大家是如何处理的?
各位兄弟 刚做滴定0.1mol/l盐酸浓度时 制备的盐酸浓度是0.09488mol/l,按照化学试剂国标要求是应在规定浓度值的加减5%范围内。按照此的话就不合适了?
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
请问各位大侠:环保部-高分辨气相/质谱法分析二恶英类的系列方法中,在前处理过程中,都提到用盐酸消解:“向样品中加盐酸,搅拌,使其与盐酸充分接触并观察发泡情况,必要时再添加,直至不发泡为止。若样品中不含碳状物时,可以活省略盐酸处理”。这里,加盐酸究竟是为什么,用消解的目的是什么?所谓碳状物究竟对后续的提取有影响,还是净化分离有影响?还是最终的上机分析测试有影响?会产生什么影响?没想明白,肯请位专家能给予指导,谢谢!
请问用什么洗涤新购买的容量瓶和移液管?拿蒸馏水冲了几次,发现挂壁很严重。用10%盐酸浸泡,效果会怎么样?10%盐酸如何配制呢?
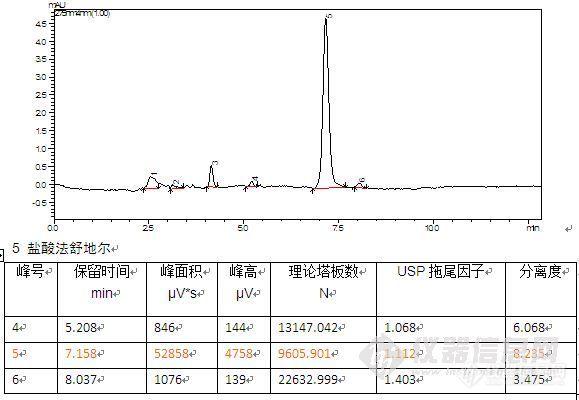
盐酸法舒地尔分析条件 流动相:1.0%三乙胺(用磷酸调pH至7.0):甲醇=50:50 流速:1.0 mL/min柱温:30 ℃ 检测器:UV 275 nm 进样量:20 μL 色谱柱: Platisil C18,250×4.6 mm,5 μm (Cat#:99503)http://ng1.17img.cn/bbsfiles/images/2012/08/201208161645_384183_2370618_3.jpg
按照国标的盐酸萘乙二胺方法测定亚硝酸盐的含量,配制盐酸萘乙二胺的时候发觉很难溶解,溶剂上会出现一些类似于沉淀的物质,放置几天后,发现瓶子底部有一层白色的类似于粉末的物质,摇匀后,溶剂呈现出浑浊的状态,这是什么原因呢?
混合碱含量测定中,用盐酸滴定溶液至橙色后,为什末将溶液煮沸,放冷却后继续滴定至溶液再次出现橙色才为第二终点?
HJ 479-2009 环境空气 氮氧化物(一氧化氮和二氧化氮)的测定 盐酸萘乙二胺分光光度法[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181815]HJ 479-2009 环境空气 氮氧化物(一氧化氮和二氧化氮)的测定 盐酸萘乙二胺分光光度法.pdf[/url]
做玩具特定元素的迁移,方法中需要使用0.07mol/L盐酸,GB 6675.4中规定盐酸的浓度范围(0.07[font=宋体][size=10.5000pt]±0.005[/size][/font])mol/L关于这个盐酸溶液,有一些疑问:1、配制该选用何种方法a、直接用盐酸配制成0.07mol/L的浓度,然后再用基准试剂标定b、配制成GB 601种列出的浓度并标定,再定量稀释成0.07mol/L2、浓度标定问题a、如果按上述a法配制,标定是否要按GB 601,俩人8平行,极差等指标也要符合GB 601的规定b、如果按上述a法配制,浓度是否可以直接按母液的标定浓度按稀释倍数直接换算个人看法:方法中规定了浓度范围(0.07[font=宋体][size=10.5pt]±0.005[/size][/font])mol/L,浓度的要求比标准滴定溶液的要低,可以按方法a配制,但是没必要俩人8平行,标定结果也没必要符合GB 601中规定的指标要求,平行3~4次标定,符合(0.07[font=宋体][size=10.5pt]±0.005[/size][/font])mol/L的要求即可。个人理解,欢迎各位大侠提出各种看法。
桑蚕丝和莱赛尔混纺,用37%盐酸法室温溶解莱赛尔纤维,能溶解桑蚕丝吗?
取浓盐酸(37%)4.5mL加水1升,得摩尔浓度约0.05mol/L盐酸标准溶液。硼砂取1g左右定溶至100mL。甲基红指示剂(由黄变红为终点)。标定时取5mL硼酸标准溶液,用盐酸进行滴定。正常消耗盐酸体积应为5mL左右,结果今天用了6mL多。重配指示剂,无效;重配硼酸标准溶液,无效;重配盐酸标准溶液约0.1mol/L(即9mL浓盐酸+1L水),并重配硼酸标准溶液取2g左右定容至100mL,依然无效!两种浓度的盐酸和两种浓度的硼砂都互滴了,还是无效;换了另一瓶未开封的硼砂重配,盐酸也换了一瓶新的开口,还是一样的结果!不知道问题是否出在了硼砂上呢?硼砂是有结晶水的,放在原试剂瓶中闭盖保存。以前也是这样用的,但一直好用。请大侠指点!
各位同仁,很久没有做盐酸标定了,昨天突然要用,然后查书看了标准,说煮沸前后滴定都是暗红色。我用无水碳酸钠,甲基红溴甲酚绿做指示剂,十滴。滴定一开始是绿色的,逐渐变紫,我滴定到玫瑰紫的时候就放电驴上煮沸冷却后再滴两滴,又变成了暗红色现在想知道的是标准上说的前后煮沸之前的暗红色是和煮沸后的暗红色程度那么深么
仪器及检测元素:AFS3000,Se溶样过程:样品消解完后,我加10ml浓盐酸(GR),样品呈墨绿色,加水定容到50ml,溶液颜色褪去,可测得的结果和直接用20%盐酸定容测得的结果相比,前者严重偏低。还有一个问题,先加浓盐酸,加标和不加标的样测得到结果基本一致,严重偏低。疑问:会不会是先加浓盐酸,使得Se挥发?
我们用的的万通 808titrando-1仪器,滴定液盐酸,测试溶液是含氨的溶液,滴定曲线有两个负的突跃,在测定有时仪器默认为第一个为终点,但我们的认为第二个是终点,现在我们要在仪器终点确认参数设置为第二个为终点(验证后实施)。现在的问题是:此滴定是否有两个突跃,其原理是什么;是否第二个突跃为终点?
目的:通过观察盐酸林可霉素对小鼠肠道菌群,肠道组织病理学改变,血中淋巴细胞数的影响规律,以期为科学研究正确使用盐酸林可霉素造成菌群失调动物模型提供参考资料。方法:盐酸林可霉素连续灌胃3d停止给药,于停药后第1天,第4天,第7天,和第10天检测肠球菌数,双歧杆菌数,血中淋巴细胞数和肠道病理改变,评价盐酸林可霉素对小鼠肠道菌群的影响规律。结果:灌胃3d停止用药后第1天和第4天双歧杆菌减少,肠球菌增加,与正常组比较差异有统计学意义(P0.05),肠道黏膜皱褶变浅,上皮内杯状细胞减少。停药后第1天出现血中淋巴细胞数减少。结论:盐酸林可霉素短期大量给药,可造成小鼠菌群失调,肠道组织损伤,免疫功能受损,该损伤持续约1周。盐酸林可霉素是科学研究中用于造成菌群失调动物模型的常用抗生素,其抑制细菌生长,尤其抑制益生菌的作用非常明显,但各家用该药的方法、剂量有较大差别,由于动物的耐受性较强,菌群失调能持续的时间不清,本实验尝试在肠道菌群变化、肠组织损伤等方面来研究林可霉素造成菌群失调的规律。以期为该药在科学研究中的使用提供数据依据,现报告如下。
瑞士万通的905自动电位滴定仪,搭配光度电极。用EDTA滴定镍钴锰溶液的总量。如果滴定溶液中不加盐酸羟胺,初始电位很低很低,突越很不明显,加入盐酸羟胺初始电位就有700多mv,正常。但是观察溶液颜色,加入盐酸羟胺前后基本无变化。这是什么原因?滴定用的是可见光波长。
5月3日上午,南昌一化工厂发生盐酸泄漏,附近居民出现头晕、呕吐等身体不适症状。事发时,该厂仓库在处理一批过期盐酸时,桶子发生破裂,造成泄漏,附近植被树叶因此出现不同程度枯萎,周边居民感觉刺鼻,头晕、呼吸困难等。常用的方法就是稀释和沙土覆盖处理,盐酸具有挥发性,你觉得这样处理妥当吗?你有啥治理办法?
盐酸的发烟温度?
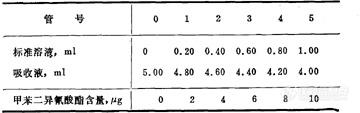
空气中甲苯二异氰酸酯的测定方法 盐酸萘乙二胺比色法 1 原理甲苯二异氰酸酯水解产生相应的芳香胺,芳香胺与亚硝酸钠重氮化后,再与盐酸萘乙二胺偶合生成紫红色,比色定量。2 仪器2.1 冲击式吸收管。2.2 抽气机。2.3 流量计,0~1L/min。2.4 具塞比色管,25ml。2.5 恒温水浴箱。2.6 分光光度计。3 试剂3.1 吸收液:量取25ml盐酸(20=1.19g/ml)与500ml水混匀,加入250ml二甲基甲酰胺,用水稀释成1L,临用前配制。3.2 亚硝酸钠-溴化钠溶液:称取3g亚硝酸钠和5g溴化钠,用80ml水溶解后,稀释成100ml。3.3 氨基磺酸铵溶液,100g/L。3.4 盐酸萘乙二胺溶液,10g/L。3.5 标准溶液:于25ml量瓶中加入5ml二甲基甲酰胺,准确称量,加入1~2滴甲苯二异氰酸酯,再准确称量,两次称量之差即为甲苯二异氰酸酯的质量,然后用二甲基甲酰胺稀释到刻度。计算1ml溶液中甲苯二异氰酸酯的含量,再用二甲基甲酰胺稀释成1ml=100微克甲苯二异氰酸酯的溶液。取5.0ml上述溶液于50ml量瓶中,加入7.50ml二甲基甲酰胺及1.25ml盐酸,混匀后用水稀释至刻度,配成1ml=10微克甲苯二异氰酸酯的标准溶液。4 采样将装有10ml吸收液的冲击式吸收管以1L/min的速度抽取50L空气。5 分析步骤5.1 对照试验:同采样,将吸收管装好吸收液带至现场,但不抽取空气,照样品分析。5.2 样品处理:采样后,用吸收管中的吸收液洗涤进气管内壁3次,由每个吸收管中各量取5.0ml样品溶液,分别放入比色管中,供测定用。5.3 标准曲线的绘制:按表2配制标准管。表2 甲苯二异氰酸酯标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201410_52373_1625938_3.jpg[/img]向各标准管中各加入0.2ml亚硝酸钠-溴化钠溶液(3.2)混匀,放置1min,再加0.2ml氨基磺酸铵溶液(3.3),激烈振摇,待气泡消失后,放置2min,加入1ml盐酸萘乙二胺溶液(3.4),充分混匀后,在20℃水浴中,放置20min,于波长560nm下比色,以甲苯二异氰酸酯含量对吸光度作图,绘制标准曲线。5.4 测定:按5.3相同的操作条件,将处理后的样品进行测定,由标准曲线上查出甲苯二异氰酸酯含量。6 计算X=2C/V0式中:X——空气中甲苯二异氰酸酯的浓度,mg/m3;C——吸收管中所取样品溶液中甲苯二异氰酸酯的含量,?g;V0——标准状况下的样品体积,L。7 说明7.1 本法的检测限为0.9?g/5ml。测定范围为2~10微克/5ml。当甲苯二异氰酸酯的浓度为2、4、6、8、10微克/5ml时,变异系数分别为7.1%、5.8%、6.5%、5.6%、5.8%。7.2 采样后样品可保存3天。7.3 吸收液应临用前配制,因久置后其中盐酸逐渐与二甲基甲酰胺作用而失去酸性。
请各位指教!用电位滴定仪标定盐酸,基准物质是无水碳酸钠,滴定过程还需煮沸吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP