求助各位:在单位用waters2695液相做左乙拉西坦,流动相:甲醇:ph=4.5的纯水=3%:97%;由于紫外响应在210nm,感觉出来的谱图基线超不平,杂质超多,血浆里杂质也多的不得了。。。。可是我的目标峰面积很小,峰型差还拖尾,感觉就是做不下去。可是领导坚持让用液相做,听崩溃的,有没有大虾有办法呢?
各位高手好! 刚学做手性不久,这两天做奥拉西坦,文献用的是大赛璐OC柱,流动相是正己烷与乙醇,我这边没有OC柱。用过正相AD,OD,OJ,IC柱,流动相用过乙醇,异丙醇,正己烷;反相AD,OD,OJ,SCA,chirobiotic T柱,流动相用过乙腈,甲醇,水;添加剂还没有试过,都没有做出来; 有没有哪位大侠做过或是相似结构的,OC柱能用我有的这些柱子配哪种流动相代替呢,请给小女子指点一二,不甚感激!!
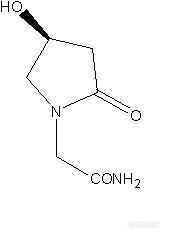
各位高手好! 刚学做手性不久,这两天做奥拉西坦,文献用的是大赛璐OC柱,流动相是正己烷与乙醇,我这边没有OC柱。用过正相AD,OD,OJ,IC柱,流动相用过乙醇,异丙醇,正己烷;反相AD,OD,OJ,SCA,chirobiotic T柱,流动相用过乙腈,甲醇,水;添加剂还没有试过,都没有做出来; 有没有哪位大侠做过或是相似结构的,OC柱能用我有的这些柱子配哪种流动相代替呢,请给小女子指点一二,不甚感激!!http://ng1.17img.cn/bbsfiles/images/2012/04/201204082229_360092_2327592_3.jpg
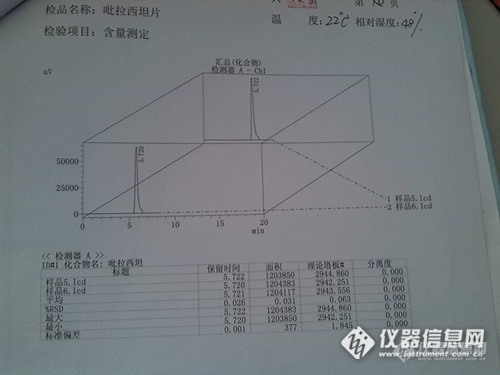
菜鸟———征服吡拉西坦片含量测定 本人是一个药品检验的新手,世称菜鸟。这是第一次做化学药品检验,在一位多年从事药品检验的师傅指导下,做了吡拉西坦片含量测定的检验。如有不妥之处请各位老师批评指正。 吡拉西坦片是化学药的一种,为白色或类白色片。其含量测定按照《中国药典二部2010版》的规定检测。正文P337。 含量测定的具体程序:取本品20片,精密称定,研细,精密称取适量(约相当于吡拉西坦0.1g),置100ml量瓶中,加流动相适量,振摇使吡拉西坦溶解,用流动相稀释至刻度,摇匀,滤过,精密量取滤液5ml,置50ml量瓶中用流动相稀释至刻度,摇匀,精密量取10ul注入液相色谱仪,记录色谱图,另取吡拉西坦对照品,同法测定。按外标法以峰面积计算,既得。色谱条件与系统适用性检查: 用十八烷基硅烷键合硅胶为填充剂;以甲醇:水(10:90)为流动相,检测波长为210nm。理论板数按吡拉西坦峰计算不低于2000。 仪器设备为岛津液相色谱,配有紫外检测器,自动进样器。这是本次检品吡拉西坦片。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291604_512123_2764104_3.jpg对照品谱图。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291607_512124_2764104_3.jpg检品谱图。检品做了两个平行,分别进了两针。http://ng1.17img.cn/bbsfiles/images/2014/08/201408291615_512131_2764104_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/08/201408291613_512129_2764104_3.jpg测得本检品含量为:97.4%。相对平均偏差:0.4%。符合药典规定。
【作者】 肇丽梅; 孙亚欣; 邱枫; 杜晓明;【机构】 中国医科大学附属第二医院临床药理研究室; 中国医科大学附属第二医院临床药理研究室 辽宁沈阳110004; 辽宁沈阳110004;【摘要】 目的:建立测定人血浆中茴拉西坦代谢物对甲氧基苯甲酰氨基丁酸(ABA)浓度的高效液相色谱方法。方法:血浆样品经醋酸乙酯提取后,以乙腈-磷酸盐缓冲液(pH3.0,30∶70)为流动相,经D iamonsil-C18色谱柱分离,251nm波长检测。结果:ABA浓度在0.1~20.0mg.L-1范围内线性关系良好,最低检测浓度为0.1mg.L-1;低、中、高3个浓度的提取回收率大于85.7%,日内、日间相对标准差低于10%。结论:本方法准确、快速,可满足临床药动学研究的要求。 【谱图】http://ng1.17img.cn/bbsfiles/images/2012/08/201208142246_383922_1609970_3.jpg
急急急!!!实验室检测的哌拉西林酸在水中的溶解度不合格,哪位亲有相关的文献资料送上,或者说说与那些因素有关,不胜感激

药物名称:奥拉西坦。货号:82006。今日抽奖结果:[align=center][img=,690,303]https://ng1.17img.cn/bbsfiles/images/2019/10/201910090929359333_5216_708_3.png!w690x303.jpg[/img][/align][align=center][img=,690,322]https://ng1.17img.cn/bbsfiles/images/2019/10/201910090929424610_1069_708_3.png!w690x322.jpg[/img][/align][align=center][img=,690,372]https://ng1.17img.cn/bbsfiles/images/2019/10/201910090929481836_8990_708_3.png!w690x372.jpg[/img][/align][align=center]=================================[color=#ff0000]活动规格[/color]====================================[/align][align=left][color=#ff0000]【活动时间】:每个工作日10:00-15:00【活动内容】:根据迪马产品资料:《药物检测应用文集》,每日会出一个化药或中药名称标题,版友根据标题找出相应迪马产品,将从回答正确者中利用抽奖软件抽取以下奖项。[/color][/align][align=left][color=#ff0000]【活动奖励】:一等奖:3个钻石(2人),二等奖:2个钻石币(3人),三等奖:1个钻石币(5个人)。[/color][/align][align=left][color=#ff0000]【注意事项】:一定要在迪马产品资料《药物检测应用文集》中找出相应迪马产品。[/color][/align]
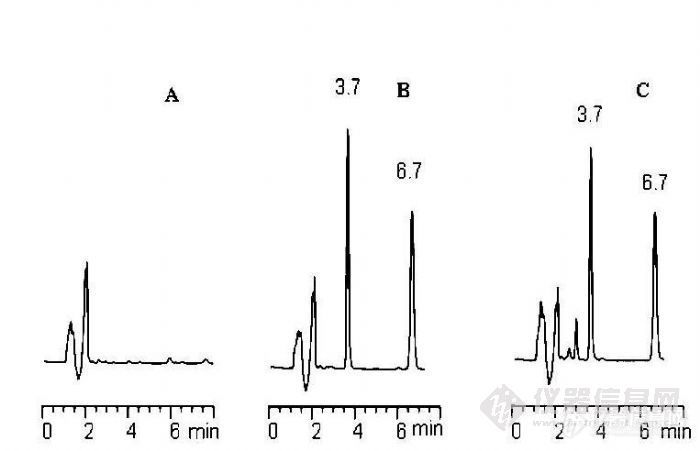
适应症脑梗塞,神经内科药理作用本品有改善记忆障碍的作用,能对抗缺氧引起的记忆减退。口服消除半衰期平均22分钟。起效快,作用强,毒性低。用于脑血管病后遗精神行为障碍,可使生活能力提高,记忆再现。无镇静作用。色谱柱信息:XB-C18 5μm,4.6*200mm;PN:ULT 5B18420;SN:210802458色谱条件暂时保密。详见附件,有很大的参考价值的哈,呵呵。。。本文主要探讨茴拉西坦有关物质前处理超声溶解时间不溶对有关物质的影响。1.超声1分钟,进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031518_455752_1621890_3.gif2.样品超声10分钟处理后进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455753_1621890_3.gif3.样品超声10分钟处理后进样色谱图:http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455754_1621890_3.gif4.不同超声时间色谱图对比。.http://ng1.17img.cn/bbsfiles/images/2013/08/201308031519_455755_1621890_3.gif5.结论:初步估计是由于超声时间较长,使其辅料溶解出来了,或是由于超声室水温身高(冰浴试验过),10分钟主峰后产生的杂质在15分钟后消失了,此杂质不稳定?抑或是此杂质再降解为其他杂质,从10分钟和15分钟色谱图中色谱峰面积之和推出,这样在物料上平衡才说的过去,头痛,具体情况没有进行再详细的研究。或是本品口服消除半衰期平均22分钟,表明本品不稳定,超声不同时间对该品稳定性有极大的影响。
求助各位大侠,对齐拉西酮怎么分析?用液相色谱的话什么柱子及条件比较好?非常重要,请各位指点http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
建立一种灵敏的检测人尿和血浆中劳拉西泮的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法。酶水解的尿和血浆样品在pH=9用乙醚提取,提取物用MTBDMS衍生化,衍生物用HP-5毛细柱进行色谱分离,用电子捕获检测器检测。尿和血浆中提取率分别为844%和813%,检测限分别为42ng/mL和24ng/mL。方法已用于口服2mg劳拉西泮人的尿和血浆分析,分析结果表明本方法适合于麻醉抢劫案中药物分析。本方法已与非衍生化、TMS衍生化[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法比较,本方法是最灵敏的方法。
酒石酸唑吡坦和右佐匹克隆互相干扰吗
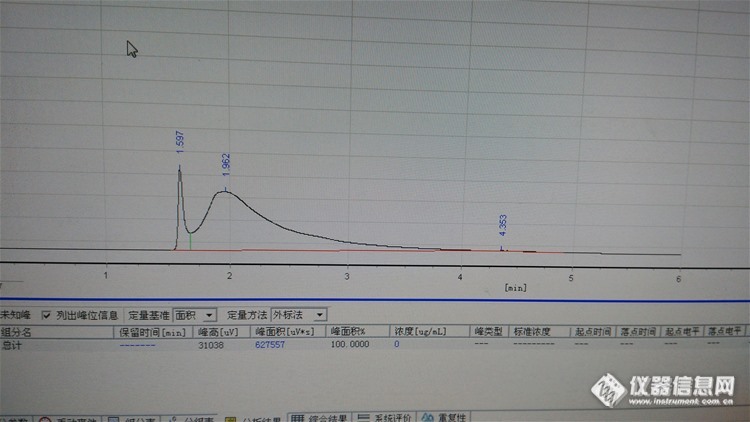
二硫化碳做解析夜分级乙酸甲酯,柱温已经降到40了,出峰还是这样~请问如何能减少二硫化碳的影响~第二张为单独纯二硫化碳峰,文件不能大于5Mhttp://ng1.17img.cn/bbsfiles/images/2015/11/201511061013_572465_3054588_3.jpg
报名链接: http://www.pharmogo.com/technical.php?act=show&id=46主 题拉西地平不同制剂的人体PK曲线预测:种属外推及Biorelevant溶出方法开发时 间2015年4月2日(周四)下午2:30-4:00主办方上海凡默谷主讲人陈涛 产品经理【内容】溶出度是固体制剂功能性评价参数,是体外评价样品质量、判断药物疗效的有效药学研究手段。开发合理的溶出度测定方法,特别是与生物体相关的溶出方法(biorelevant dissolution method),对于准确评价药物在人体内的吸收和处置过程、预测人体的药代动力学曲线将起到至关重要的作用,进而为制剂的处方开发提供有效的指导。本次网络会议,将选择典型的BCS II类药物-拉西地平作为模型药物,通过GastroPlus软件的机制性吸收和处置模型(ACAT and PBPK model)建立该药物在狗及人体内的PK模型;然后借助GastroPlus独特的Z-facter溶出模型,筛选biorelevant体外溶出方法,并进一步预测该药物不同处方在狗及人体内的PK曲线,为新制剂的开发提供指导。通过此次网络会议,您将详细地了解Z-facotr模型在筛选体外溶出方法、评价不同制剂处方中的重要作用;并进一步熟悉GastroPlus的机制性模型在体内PK曲线预测、种属外推等方面的应用。将拓展您对GastroPlus软件在制剂研究中的应用认识,指导您相关的科研工作!

各位好!有没有做过头孢替坦酸的?按照日本药典方法做头孢替坦酸有关物质实验的时候主峰前面出现了一个小峰,谱图如下:http://ng1.17img.cn/bbsfiles/images/2015/08/201508051144_559190_1866875_3.jpg实验条件:检测器:VWD;检测波长:254nm;色谱柱:C18色谱柱(L=250mm,5μm,φ=4.6mm);0.1mol/L磷酸溶液:取11.53g磷酸加水1000ml,摇匀,即得;流动相:0.1mol/L磷酸-色谱甲醇-色谱乙腈-色谱冰乙酸=1700:105:105:100。(此流动相临用新制)流速:2.0ml/min;柱温:40℃。样品前处理:取甲醇10ml,注入10μl于液相色谱仪。如果样品溶解用甲醇和水的话前面的峰就没有了,不知道什么情况。求各位大侠支招!
括号内为每个品种注册排序(只收集每个品种前3家)。厂家2008-2014年获批的品种2011-2014年申报的品种正大天晴恩替卡韦(1),替加环素(2),伊马替尼(1),达沙替尼(1),卡培他滨(3),地西他滨(1),帕洛诺司琼(2),比阿培南(2),瑞舒伐他汀(3),雷替曲塞(1),阿折地平(1),米格列奈(1),巴洛沙星(2)普拉曲沙(1),坦罗莫司(1),托法替布(2),泊沙康唑(2),匹杉琼(2),阿比特龙(1),来那度胺(2),福司氟康唑(1),卡博替尼(1),索利那新(2),氟维司群(1),巴多昔芬(3),阿哌沙班(1),硼替佐米(3),地特胰岛素(2),聚乙二醇干扰素α-2a(1),多立培南(1),度他雄胺(3),安立生坦(2),利奈唑胺(3),米铂(3),罗米地辛(1),西那卡塞(3),阿福特罗(1),阿加曲班(3),右兰索拉唑(2),达非那新(1),多黏菌素E(1),阿瑞匹坦(3),福沙匹坦(1),塞来昔布(2),阿格列汀(2)豪森药业替加环素(3),伊马替尼(2),地西他滨(3),依替巴肽(1),头孢地尼(3),米格列奈(3)达比加群酯(1),舒尼替尼(3),索拉非尼(1),匹杉琼(1),卡格列净(1),帕唑帕尼(3),芬戈莫德(3),鲁拉西酮(1),来那度胺(1),阿昔替尼(3),维格列汀(1),泊马度胺(1),阿哌沙班(3),硼替佐米(1),安立生坦(1),利奈唑胺(2),厄洛替尼(2),达沙替尼(2),右兰索拉唑(1),福沙匹坦(3),利伐沙班(2),米卡芬净(1),地加瑞克(1),阿考替胺(1),瑞替加滨(3),地拉罗司(1),帕利哌酮(1),维拉佐酮(1),普卡必利(2),依折麦布(1),伊潘立酮(1),比伐卢定(2),布南色林(3)恒瑞医药右美托咪定(1),非布司他(3),卡培他滨(2),左亚叶酸钙(1),塞来昔布(1),培门冬酶(1),替吉奥(2)磺达肝癸钠(1),替格瑞洛(1),帕瑞昔布(2),阿齐沙坦(1),托伐普坦(1),卡泊芬净(1),度他雄胺(1),吉非替尼(2),苯磺贝他斯汀(2),贝美前列素(1),他达拉非(1),伊伐布雷定(1),索利那新(1),利马前列素(2),氯维地平(3),钆特酸葡胺(1),培化干扰素α-2b(2)齐鲁制药卡培他滨(1),帕洛诺司琼(1),替吉奥(3),左西孟旦(1),氨磺必利(1)达比加群酯(2),索拉非尼(2),舒尼替尼(1),帕唑帕尼(1),帕瑞昔布(3),阿昔替尼(2),卡巴他赛(3),阿哌沙班(2),厄洛替尼(1),吉非替尼(1),利伐沙班(1),西地那非(2)石药集团厄他培南(1),比阿培南(3)替格瑞洛(2),索拉非尼(3),舒尼替尼(2),决奈达隆(3),硼替佐米(2),吉非替尼(3),达沙替尼(3),伊伐布雷定(2),利伐沙班(3),西格列汀(2),头孢唑兰(1),泰比培南酯(1)南京华威0匹杉琼(3),比拉斯汀(1),芬戈莫德(1),阿齐沙坦(2),决奈达隆(2),托非索泮(2),伊曲茶碱(1),维格列汀(2),米铂(2),西那卡塞片(1),瑞替加滨(2),普卡必利(1),碳酸司维拉姆(2),雷美替胺(1),艾曲泊帕(1),罗卡色林(1),艾司利卡西平(3),咪达那新(1),米诺膦酸(2)天津汉康0比拉斯汀(2),加雷沙星(1),阿塞那平(3),阿伐那非(1),博舒替尼(2),罗氟司特(1),鲁拉西酮(3),瑞替加滨(1),伊潘立酮(2),布南色林(1),雷美替胺(2),艾司利卡西平(1),咪达那新(2),米诺膦酸(3),克利贝特(2)科伦药业艾司西酞普兰(2)[
各位大佬,有没有分析过碳酸二甲酯,碳酸二乙酯。 碳酸甲乙酯的分析条件。 什么色谱柱。 MS还是FID检测器。大佬能给个建议么?
天美7890二气相,填充柱 柱温55,进样器120,检测器160乙酸乙酯浓度 (微克/毫升 ) 面积45.05, 2180590.1 46447180.2 101218曲线y=590.5x-5580.5 r=0.9994活性炭管质控样 加1毫升二硫化碳解析30分钟,进样1微升,结果只有一半多一点问:1.活性炭管保存方法?证书写室温,我就没放冰箱。乙酸乙酯只有半年,做时快到有效期了,可能也有影响吗? 2.解析效率怎么做?质控样给没有空白活性炭管,可以用家里的空白活性炭管吗,但他和买的管不是一批呀?但不做解析效率,解析能100%吗恳切希望做过的前辈指点!
活性炭管同时采了甲苯与乙酸甲酯,二硫化碳解吸。甲苯我一直用非极性柱做,乙酸甲酯极性柱做。这要分两次进样麻烦。岛津2014c自动进样那台装的是OV101非极性柱。我想用OV101柱(25m*0.20mm*0.25um)同时做甲苯和乙酸甲酯。最大的麻烦是二硫化碳与乙酸甲酯分离的问题,有谁成功的用非极性柱分离过二硫化碳溶剂峰和乙酸甲酯吗?
PC(碳酸丙烯酯),EC(碳酸乙烯酯),DMC(碳酸二甲酯),DEC(碳酸二乙酯),EMC(碳酸甲乙酯)混合物可以用GCMS 测试吗?还是GCFID

前几天同事扩项工作场所空气中饱和脂肪酸酯类物质包括乙酸甲酯,乙酸乙酯,乙酸丙酯,乙酸丁酯,标准是GBZ/T160.63-2007。柱子是SH-Rtx5(30m*0.32mm*0.25um),同事欲恒温同时分离这四种酯类,我提示乙酸乙酯,乙酸丙酯,乙酸丁酯混一起恒温做没事,如果乙酸甲酯也混一起做那么会与溶剂二硫化碳峰难分离,于是他计划先做乙酸乙酯,乙酸丙酯,乙酸丁酯再另外单独做乙酸甲酯。 乙酸乙酯,乙酸丙酯,乙酸丁酯色谱条件:岛津气相色谱GC2010PLUS 柱温60℃ 检测器进样器均为200℃ 分流比50 恒线速度22cm/shttp://ng1.17img.cn/bbsfiles/images/2017/01/201701191702_673938_2103464_3.jpg三种乙酸酯峰型还不错。接下来他说想试试乙酸甲酯在这个条件下出峰会怎么样?因为我之前用OV101做过二硫化碳中乙酸甲酯,它是紧挨着在二硫化碳前出峰,同时也用DB-FFAP做过它是在二硫化碳之后。SH-Rtx5极性比OV101强些 比DB-FFAP弱很多,那么在二硫化碳之前还是二硫化碳之后出峰呢? 看到甲乙丙丁突然有了一想法:不是有碳数规律吗?利用碳数规律推测乙酸甲酯的保留时间:保留时间:乙酸乙酯 2.854min 乙酸丙酯 3.562min 乙酸丁酯5.162min 二硫化碳2.612min碳数规律:lnt‘=An+C t’为调整保留时间 n为同系物中碳个数 A, C均为常数首先精确计算死时间:间隔均匀同系物 精确死时间计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616020890_01_2103464_3.pngtm=(2.854*5.162-3.562*3.562)/2.854+5.162-3.562-3.562=2.292min调整保留时间代入碳数规律公式:乙酸乙酯ln0.562=4A+C 乙酸丙酯 ln1.27=5A+C 求得乙酸甲酯调整保留时间lnt’=3A+C t’=0.249min乙酸甲酯的预测保留时间0.249+2.292=2.541min.这个保留时间在二硫化碳(2.612min)之前,两者仅仅相差0.07min。于是预测同条件下乙酸甲酯在二硫化碳之前出峰并且分离度不好!同条件实验做二硫化碳中乙酸甲酯3000ug/ml来验证:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616132954_01_2103464_3.jpg实验结果乙酸甲酯保留时间是2.571min与预测的2.541min比较符合,外推是有误差的,而且本例碳数不多,碳数多些会更准确。降低柱温至40℃,线速度15cm/s 乙酸甲酯与二硫化碳分离达到定量要求!http://ng1.17img.cn/bbsfiles/images/2016/09/201609061616_608604_2103464_3.jpg 结论:碳数规律还是比较准的,可以预测出峰情况。
碳酸二苯酯用液相色谱法怎么做啊,日本食品卫生法里那个流动相浓度斜率从3:7到100:0直线斜率进行35min,用乙腈冲洗10min是怎么设置的啊。碳酸二苯酯液相色谱法还有别的方法么。
我们公司是做改性塑料的,需要大量的活性碳酸钙填料,而活化度是表征碳酸钙活化程度好坏一个重要的指标,根据《HGT 2567-2006 工业活性沉淀碳酸钙》和《HGT 3249.3-2008 塑料工业用重质碳酸钙》中关于活化度的规定来看,都是采用国标《GBT 19281-2003 碳酸钙分析方法》中规定的方法来测的,但是国标中针对一些测试的细节没有描述。具体方法见附件,但是在测试的过程中发现有部分供应商的产品容易发生团聚现象,给活化度定量带来极大误差,想请教专业人士,我司所用方法是否有误?产生这个团聚现象的原因是什么?如何避免?有做碳酸钙的朋友也可以发表下意见,谢谢!
碳酸二苯酯用液相色谱法怎么做啊,日本食品卫生法里那个流动相浓度斜率从3:7到100:0直线斜率进行35min,用乙腈冲洗10min是怎么设置的啊。碳酸二苯酯液相色谱法还有别的方法么。
分离活性炭管中苯、丙酮、乙酸乙酯,FID检测器,FFAP柱(30*0.32*0.50),用二硫化碳做溶剂,请问能用用同一个条件:进样口温度150℃,检测器温度180℃,柱温40℃,以5℃/min升至80℃。把物质分离出来吗?谢谢
萤石中碳酸钙怎么做?
我公司做循环冷却水中的铜离子测定?根据国家标准《GBT 13689-2007 铜的测定》要用四氯化碳进行萃取!!!可四氯化碳毒性太大了,打算尝试用乙酸丁酯代替之。在这里乙酸丁酯能代替四氯化碳作萃取剂嘛?望高手解答。如有关于有机萃取剂的资料 也希望能共享下。
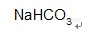
【中文名称】碳酸氢钠;酸式碳酸钠;重碳酸钠;小苏打;焙烧苏打;重碱【英文名称】sodium bicarbonate;sodium hydrogen carbonate【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203072013_353130_1855403_3.jpg【相对分子量或原子量】84.01【密度】2.20【毒性LD50(mg/kg)】 大鼠经口4300【性状】 白色单斜晶体。【用途】 是重要的常用药物(消化剂、制酸剂),又是制灭火剂、焙粉和清凉饮料等的原料。还可用于饲料作饲料添加剂的缓冲剂。【制备或来源】 可由碳酸钠浓溶液或结晶碳酸钠吸收二氧化碳而制的。是氨碱法制纯碱的中间产物。【其他】 在热空气中,能缓缓失去一部分二氧化碳,加热至270℃失去全部二氧化碳。
[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]中,用碳酸做淋洗液,与用KOH做淋洗液有什么区别
用戴安的离子色谱,最近想分离溴酸根和氯离子,淋洗液是碳酸钠和碳酸氢钠,试了不同的配比都无法分开,想单独用碳酸氢钠做淋洗液不知道可行吗?
领导要求做碳酸氢钠的X衍射,想知道碳酸氢钠的晶型,但没有相关的操作规范和标准,哪位前辈有相关资料,请指点下,在线等....







 我要推广仪器
我要推广仪器
 下载APP
下载APP