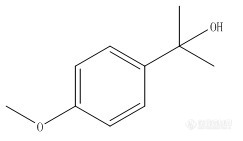
这种苯基异丙醇易于形成脱水的分子离子峰么,大家有类似的谱图么?谢谢!http://ng1.17img.cn/bbsfiles/images/2012/12/201212221329_414443_1618372_3.png
本人最近在做溶剂残留,用HP-INNOWAX色谱柱,今天将异丙醇、正丁醇、二甲苯配在一起,却怎么也分离不出正丁醇和对二甲苯了!!真是着急!请教各位高手,做溶剂残留,用HP-INNOWAX色谱柱时,如何设计合适的程序升温,才能有效的把醇类和苯类分开来呢?谢谢拉!!
[color=#444444]想知道反应中有无丁醇生成,溶剂为甲苯,可以用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]检测出丁醇吗?多谢啦[/color]
测四氟丙醇中的杂质叔丁醇的含量。因为现有两台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC979/GC1690,手动进样0.8UL,都是FID。柱子都是兰化所的1701毛细柱。分析同一个样品,总峰面积1690为1080W,9790的为998W,结果1690那台叔丁醇的含量是9790的两倍,做了3次试验都是一样情况。两台色谱的载气,空气,氢气的流速都一样。不知道怎样才能确定哪个是准确的。是不是用内标?本人接触色谱不久对内标不是很懂,对叔丁醇选什么内标物好?内标物是不是要特别买?我看试剂的含量都是个范围。
DB-624能将苯和异丁醇分开吗?如果能的话具体的程升是怎样的?进样口,流速,检测器各是多少?谢谢
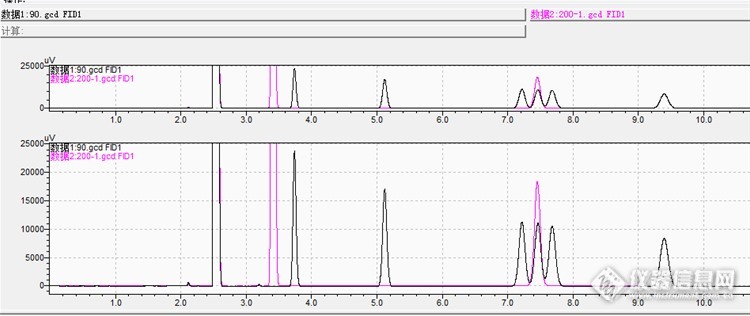
空气与废气样品中挥发性有机物常见的分析方式是活性炭管采样二硫化碳解吸。比如苯系物,丙酮,丁酮,环己酮,正己烷,乙酸甲酯,乙酸乙酯,乙酸丁酯,二氯甲烷,1,2-二氯乙烷,异丙醇,正丁醇。参考的依据有 环境空气苯系物的测定 活性炭吸附/二硫化碳解吸-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法HJ584-2010,《合成革与人造革工业污染物排放标准》VOCs监测技术导则 GB 21902-2008 附录C, 还有工作场所空气有毒物质测定 GB/T160 系列。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]多组分分析时尽量使用相同的条件,这样可以尽可能的减少进样而减少分析时间。这次遇到活性炭管样品要求检测甲苯,二甲苯,正丁醇,实验中发现平常的分析条件正丁醇与对二甲苯出峰时间重叠,于是通过摸索分析条件最终成功摸索出低温恒温,高温恒温,程序升温三种分离方法。 平常苯系物分析色谱条件:岛津GC-2010plus带AOC-20i自动进样器,DB-WAX(30m*0.53mm*1.0um),进样口200℃,检测器200℃,柱温恒温80℃,线速度25cm/s,分流比20,进样1ul。正丁醇采用相同的条件。苯系物与正丁醇比较图:黑色为苯系物其中三连峰是乙苯,对二甲苯,间二甲苯,红色为正丁醇与对二甲苯几乎完全重合。[img=,690,357]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281523_01_2103464_3.png[/img] 这个重叠峰分离有些特殊,因为正丁醇前面连着乙苯,后面连着间二甲苯。想分离前移或后移正丁醇峰比较大的距离才有用,之前我用极性柱DB-FFAP分离过二硫化碳中1,2-二氯乙烷与乙酸丁酯,分离异常艰难,所以刚开始我就想这个难度不小,有人说得用60m的DB-WAX,可惜我没有!当然这种分离可以换非极性柱来做,不过我不想换非极性柱,因为平常大多用这根柱子,这次想要发挥这根柱子的潜力。 虽然样品没要求测乙苯,但根据经验乙苯与二甲苯经常共存,于是配制苯,甲苯,乙苯,对二甲苯,间二甲苯,邻二甲苯 ,正丁醇约30ug/ml 混合二硫化碳溶液,另配制正丁醇二硫化碳溶液单标做定性。以下图谱因为节省分析时间只要 乙苯,对二甲苯,间二甲苯,正丁醇出峰 就停止采样,烘烤色谱柱至邻二甲苯出峰再进行下次分析。首先想到降低柱温,降低柱流速来实现分离。分析条件1:柱温改成65℃,柱流速改成17cm/s(以下各条件都是这个流速),发现正丁醇后移与间二甲苯重叠:[img=,690,287]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281443_01_2103464_3.png[/img]这说明这招有效,于是分析条件2:继续减低柱温至55℃:发现正丁醇跑到间二甲苯后面完全分离![img=,690,281]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281448_01_2103464_3.png[/img]峰是分离开了,可是正丁醇出峰时间达26min感觉太慢了,于是想加快分离速度,分析条件3:柱温改成60℃[img=,690,319]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281454_01_2103464_3.png[/img]可见正丁醇与乙苯,对二甲苯,间二甲苯完全分离,正丁醇出峰时间减少至21.5min。这个条件为低温恒温。至此分析条件摸索好了,可是再琢磨下:既然正丁醇降温可以后移,那么升温应该会前移,前移比较多的话可以跑到乙苯前面,这样出峰时间更少,岂不是更好!条件4:柱温改成90℃,这次正丁醇确实是前移了不过和乙苯重叠了四连峰变成了三连峰:[img=,561,355]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281459_01_2103464_3.png[/img]分析条件5:柱温升至100℃,正丁醇跑到乙苯前面完全分离,分析时间大大缩短!此条件为高温恒温。[img=,633,381]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281501_01_2103464_3.png[/img]想到[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]大多使用程序升温那就用程序升温再试试。分析条件6:初温50℃以3℃/min至80℃保持5min,正丁醇与间二甲苯重叠:[img=,690,338]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281507_01_2103464_3.png[/img]分析条件7:初温40℃保持2min以2℃/min升至70℃保持17min,正丁醇跑到间二甲苯后面完全分离!此条件为程序升温。[img=,690,329]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281511_01_2103464_3.png[/img]至此摸索出三种条件都可以分开乙苯,对二甲苯,间二甲苯,正丁醇,分别是低温恒温,高温恒温,程序升温三者的比较:[img=,690,309]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281515_01_2103464_3.png[/img]可见灵敏度是:高温恒温>程序升温>低温恒温 分析时间是:程序升温>低温恒温>高温恒温样品分析:因为样品可能有别的有机物,分析时间不宜太快于是采用程序升温方法,三个峰分别是乙苯,对二甲苯,间二甲苯,而未检出正丁醇:[img=,680,362]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281519_01_2103464_3.png[/img]总结:1.原因分析是通过降低柱温使得DB-WAX与正丁醇相互作用力(主要是偶极力)加强的更显著,所以与乙苯,对二甲苯,间二甲苯比较是后移,反之亦然。 2.摸索出三种方法:低温恒温,高温恒温,程序升温,灵敏度是:高温恒温>程序升温>低温恒温 分析时间是:程序升温>低温恒温>高温恒温(比较到四连峰最后一个峰的出峰时间为止)。低温恒温的好处是基线平整,高温恒温的好处是分析时间非常快,如果样品比较干净是比较适合的,而程序升温适合的样品复杂些。[u][/u]
今天实验发现 DB-WAX(30m*0.53mm*1.0um)正丁醇与对二甲苯重叠,有分开过的吗?与间二甲苯也不能重叠
正丁醇和二甲基苯分不开,要求分好以后有四个峰,最好是填充柱.
正丁醇和丁醇是一样的吗?
现做一原料残留溶剂方法验证,以DMF为溶剂,检测异丁醇、甲醇、乙腈、二氯甲烷、环己烷和甲苯。Vial Temp.: 80℃Loop Temp.: 100℃Transfer Line Temp.: 120℃Vial Equilibration Time: 30 minPresurizing Time: 2.0 minLoop Fill Time: 0.5 minLoop Fill Time: 0.05 minInject Time: 0.05minInjection Port: 140℃N2: 2.5mL/min升温程序初始40℃5min然后10℃/min升温至240℃保持2min问题:1. 空白也就是DMF中在甲醇和异丁醇出峰位置有峰,干扰样品测定,且S/N竟然已经大于了10,不能忽略不计了,怎样才能消除此干扰2. 顶空进样瓶中加入1mL液体,连续进样6针对照,RSD不好,尤其是异丁醇远远大于10%,且峰面积越来越大。什么原因呢?随着时间推移不断进对照峰面积都是逐渐增大(换过衬管和垫片,是否可以排除残留问题)在线坐等高手
请问a-苯基甲醇-乙酸指示剂如何配制fjcgx@126.com谢谢[em61] [em61]
DMA做溶剂,却有醇峰,定位炔丁醇和混合溶剂跑出来的峰,混合溶剂里炔丁醇附近有两个峰,是反应了吗
慕尼黑上海分析生化展,迪马科技最新推出了Inspire 苯基系列色谱柱,一直忙未给大家及时普及,今天来说说第一款Inspire PFP ( 五氟代苯基) 是Inspire 液相色谱柱家族新成员,针对分离极性化合物过程中的保留时间和分离度问题而特别设计。Inspire PFP 凭借其优异的选择性,可为极性化合物、复杂天然产物、位置异构体和其它相关化合物在C18 和C8 色谱柱上的分离提供一个替代和补充。Inspire PFP 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式,并具有多种作用机理,因而能够同时分离检测不同极性化合物的混合物,为目前难以解决的复杂极性和亲水性样品的分离分析提供了强有力的工具,可轻松解决其它色谱柱面临的分离难题,为用户实现强极性分析物的优异选择性提供一种更加便捷的途径。同时也为色谱工作者使用简单流动相,避免使用极端pH 条件和准备复杂流动相提供了可能性。Inspire PFP 色谱柱特点• 五氟代苯基硅烷键合在高纯硅胶基质上• 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式• 对极性化合物具有独特的保留能力• 良好的峰形、超高的柱效、分离度和使用寿命• 适用于芳环类化合物或长共轭体系化合物的分离• 优异的批次重现性增强位置异构体分离能力官能团位置的微小差异可以极大的影响分子性能,在许多情况下,传统的C18色谱柱根本无法扑捉到这种细微的差异。然而,Inspire PFP的多功能选择性却可以区分由于分析物内部微小位置变化而导致的分析物的空间位阻变化还是分析物的偶极矩偏移。色谱柱如图所示 规格 150 × 4.6 mm, 5 μm 流动相0.1% 甲酸乙腈溶液:0.1% 甲酸水溶液 = 40:60 流速1.5 mL/min 柱温室温 检测器 UV 254 nm 样品1. 3,4-二甲氧基苯酚 2. 2,6-二甲氧基苯酚3. 3,5-二甲氧基苯酚4. 2,6-二氟苯酚5. 2,4-二氟苯酚6. 2,3-二氟苯酚7. 3,4-二氟苯酚8.3,5-二甲基苯酚9.2,6-二甲基苯酚10.4-氯-3-甲基苯酚11.4-氯-2-甲基苯酚12.3,4-二氯苯酚13.3,5-二氯苯酚http://www.dikma.com.cn/u/image/2016/09/06/1473147613188048.jpg苯氧酸类化合物分子上卤素的加入可以从根本上增强化合物的极性,而极性的变化通常伴随着反相色谱柱在保留时间和分离能力上困难的增加。此时使用InspireTM PFP 是解决保留问题的最有效的方法。InspireTM PFP利用偶极-偶极和氢键作用更好地保留,区分和分离极性卤化化合物。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:0.1% 甲酸水溶液 = 50:50流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 苯氧乙酸2. 邻氯苯氧乙酸3. 对氯苯氧乙酸4. 2,4-二氯苯氧乙酸5. 2,4,5-三氯苯氧乙酸6. 2,4,5-三氯苯氧丙酸http://www.dikma.com.cn/u/image/2016/09/06/1473147817102957.jpg类固醇通过整合偶极-偶极、π-π和氢键机理,InspireTM PFP实现标准反相条件下极性化合物的最佳分离。色谱柱 如图所示 规格 150 × 4.6 mm, 5 μm 流动相甲醇:水 = 60:40 流速1.5 mL/min 柱温室温 检测器UV 254 nm 样品1.泼尼松龙3.地塞米松5.氢化可的松21-乙酸酯7.可的松-21-乙酸酯2.泼尼松4.皮质酮6.11-α羟孕酮8.11-酮孕甾酮http://www.dikma.com.cn/u/image/2016/09/06/1473148006619700.jpg甲基苯乙酮异构体目标分析物上的基团位置变化可以影响化合物的偶极矩,这种变化可以很容易被高电负性的氟原子和其它保留机理察觉,因此InpireTM PFP可以有效地用于分离甲基苯乙酮的位置异构体。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相甲醇:水 = 50:50流速1.0 mL/min柱温室温检测器UV 254 nm样品1. 邻 -甲基苯乙酮2. 对 -甲基苯乙酮3. 间 -甲基苯乙酮http://www.dikma.com.cn/u/image/2016/09/06/1473148212903667.jpg核苷酸和核苷色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相0.1% 甲酸水溶液流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 胞嘧啶2. 5'-CMP3. 5'-UMP4. 5'-GMP5. 尿苷6. 胸腺嘧啶 http://www.dikma.com.cn/u/image/2016/09/06/1473148405914511.jpg抗胃酸药色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:20 mM 磷酸氢二钾(pH 7.0) = 20:80流速1.0 mL/min柱温室温检测器UV 220 nm样品1.法莫替丁2.西咪替丁3.尼扎替丁4.雷尼替丁 http://www.dikma.com.cn/u/image/2016/09/06/1473148597658988.jpg氧化应激标记物色谱柱 如图所示规格 150 × 4.6 mm, 5 μm[/
小弟在做的溶剂标样:分别有甲醇,乙醇,异丙醇,乙酸乙酯,正丁醇,丁酮,甲苯,甲醚,甲基环乙烷,正丙酯,丁酯。。做标样的时候是这11种,但是出峰只出来9个,没有正丁醇和甲醚。。。然后单独进样的时候,正丁醇,正丙酯,甲醚的出峰时间很接近,用三种混的时候进样,却只有一个峰。不明白为什么进了三种只出了一个峰,是分离不开还是什么问题???柱温是70度。。检测器进样器都180度,毛细管柱50*25*25。。请各位大神帮忙..怎么分辨两种物质性质很相近?比如正丁醇,正丙酯,甲醚这三种为什么会那么近?
[em0812]我新买回PEG-20M色谱填充柱(3m*3mm)老化后,进正丁醇样后发现正丁醇与异戊醇很难分离,有时还被正丁醇峰覆盖了,我选择的条件是:柱温110,进样器180,检测器180。后来我又改变了条件:柱温(80-110)、进样器(145-180),又调整载气(N2),还是分离不出异戊醇,各为高手有何解决办法?[em0808][em0810][em0811]
有机化工产品精馏过程副产有丁醇水溶液,取样发现取样瓶内很快就分层了,除了水,丁醇外,还有少量甲醇杂质。[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]有啥办法准确定量其中的丁醇含量。
国内不生产 α-萘酚苯基甲醇 指示剂?
哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
配了几个异丁醇的标液,溶剂甲醇,10000ppm,5000ppm,1000ppm,但打出来不成线性关系,请问有做过的吗?还是异丁醇配置需要什么注意的地方,谢谢!
薄层色谱法1 应用范围本方法适用于眼部化妆品及卸装品中防腐剂硫柳汞和苯基汞的定性测定。2 原理样品经浸提处理后,置检液于硅胶板上,经与双硫踪络合后,用己烷-丙酮液展开,根据与标准的Rf 值比较进行定性。3 试剂3.1 0.004%双硫腙氯仿溶液(1)。3.2 展开液:己烷十丙酮(90+10)。3.3硫柳汞标准溶液:称取相当含汞1.000g(2)的硫柳汞,溶于95%乙醇十水(1+1)溶液,补加95%乙醇十水(1+1)至100ml刻度。此溶液含汞10.0mg/ml。取一定量此溶液,用95%乙醇稀释至含汞0.35mg/ml的溶液。3.4苯基汞盐标准溶液:称取相当含汞1.000g的苯基汞盐于95%乙醇十水(1+1)溶液中,并稀释至100ml刻度,此溶液每毫升含汞10.0mg。取一定量此溶液用95%乙醇稀释至每毫升含汞0.35mg。4 仪器4.1 层析缸。4.2 高效硅胶薄层板:带富集区,Merekl3727或13728或等效物。5 分析步骤5.1 样品处理取5g样品分散于15m1 95%乙醇中,超声匀浆15min,用中速滤纸过滤,滤液在水浴锅上蒸发至近干,将残渣溶于lml 95%乙醇。5.2 分别取2.0μ1检液和标准点样于硅胶薄层板的富集区,每次1μ1。用一玻璃盖覆盖在硅胶薄层板上,但勿将富集区覆盖。将双硫腙氯仿溶液喷布在富集区上,如检液含有汞化合物,则与双硫腙形成络合物,将薄层板置于装有20ml展开液的层析缸内,盖上缸盖进行层析展开。展出液前沿移动6cm以后取出薄层板,观察橙色斑点的 Rf 值并与标准点进行比较,以判定检样中是否含有硫柳汞或苯基汞(4),以及其含量是否超过化妆品卫生标准限定最大用量(5)。
哪可买到色谱纯正丁醇,异丁醇,异戊醇,2-甲基丁醇,正丙醇标样?
请教各位大神,用啥柱子能把2-甲基丁醇和3-甲基丁醇在气相色谱图中分开呢?目前我用的是安捷伦 DB-WAX柱,柱子截过几次,两种醇的特征峰重叠了,无法区分。
我们样品中要添加正丁醇。所以我们正在分析PTG(聚四亚甲基醚二醇)中正丁醇的含量,一般含量在1200ppm左右。我们分析方法是这样的:用的是内标法,内标物是正丙醇,溶剂是环己烷,内标物的配置是5ml的正丙醇用环己烷稀释到2000 .称取样品2g左右,移取20ml的内表液。摇匀,进样0.2ul.分离也很好,但是重现性很不好。同一个人同一个样品上午和下午作有时能相差200ppm.我现在怀疑环己烷是非极性溶剂,而正丁醇是极性的,环己烷能不能萃取出正丁醇?不知道各位大虾有没有什么好的溶剂溶解PTG萃取出正丁醇?还有内标法对进样量好像要求不是很大,那内标法中有哪些因素影响重复性?很急!!!!!
大家好: 一直以来 3-甲基-1-丁醇,3-甲基-2-丁醇的分离问题一直困扰着我,哪位高手能帮忙解决一下,请问用什么样的毛细柱分离,在什么条件下?先谢谢各位了。
请教三苯基膦和三丁基膦如何溶于水中? 在水中稳定性如何?毒性大吗?谢谢!
分析二甲基丁醇,三甲基丁醇,样品浓度大约为99%。请教分析方法及使用何种色谱柱?谢谢了!![em58]
国标乳品中碘的测定中,在硫酸条件下碘与丁醇反应生成丁醇与碘的衍生物,到底此衍生为何物?
各位大虾,有没有人用过五氟苯基柱的?我用的五氟苯基柱使用寿命比C18短的多了,这里有没专门做填料的告诉我原因?使用过程中是50的ACN和0.05N的PH7PBS,用完后90水冲然后甲醇冲,维护的没有问题,大概进了2000针以后塔板数从10000降到 3000,虽然2000针也到了寿命了,但和C18比起来似乎短了点.我是这样理解的,五氟苯基柱键合的时候是C4链一端连到硅基上,另一端连到五氟苯基上,由于碳链太短所以水容易侵入到硅基质,造成固定相的流失?我想知道,五氟苯基柱在键合的时候封端了吗(phenomenex)?该柱子在高水(90%)时固定相会流失吗?谢谢大家给我点建议吧
正丁醇部位的分离是天然药物化学专业分离较难的部分,但分到有价值的东西几率较大,所以值得我们为之一搏。 一般来说,正丁醇部位常含有单糖、二糖等小分子糖,多种酚类化合物,以及苷类化合物,极性较大,在硅胶柱上吸附较多,成点性差,分离效果不好。因此,一般说来,对于正丁醇部位可采取以下方法: 1. 大孔树脂柱砍段。 样品水溶解后上样,依次用水,30%、60%、95%乙醇溶液洗脱,分别合并、收集。一般说来目标化合物多在60%段,水,30%段多为一些水溶性单糖、二糖及多酚类化合物,可以考虑弃去。而95%部分多和乙酸乙酯部位大极性段重叠,可以乙酸乙酯部位合并处理。 2. 反相柱分段。 如果正丁醇部位量不是太大,或者课题组有大反相柱,可以考虑用反相柱砍段。本课题组就有一根500g的反相柱,专门用于砍段,效果比正相柱好了许多。唯一需要提醒注意的是,由于我们一般是用正相硅胶板检测化合物分离情况,所以反相柱 砍段后往往各组份在硅胶板上看起来比较混乱,不如硅胶柱砍段后那么直观,一定要小心对比,否则会越分越乱。 3.正相柱分配色谱层析。 如果正丁醇部位量太大,或者课题组没有大的相柱用来分段,那么可以考虑氯仿:甲醇:水 体系来砍段。我一般用8:2:1、7:3:1以及6.5:3.5:1依次洗脱,效果不会很好,但两到三次反复上柱后各部分还是可以看得到点的。这时再会反相柱细分,拿十到二十个点应该没问题的。 总体来说,这部分较难,但等你分到好东西了,难也值得,小小经验和大家分享,祝大家实验顺利!
求丁醇的国标 感谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP