春节好 求助氮氧自由基哌啶醇含量化验方法 谢谢!
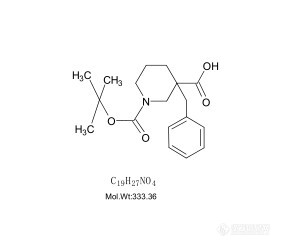
色谱世界的各位大侠们,1-boc-3-苄基哌啶甲酸文献报道说用4.6mm*150mm C18的二氧化硅柱子,用乙腈、水、三氟乙酸作为流动相,在214和254处有吸收峰。我们用紫外全波长扫描后,发现只有在204处有紫外吸收峰,可是我们用乙腈和水作流动相,这个东西在214处不出峰。这个东西该怎么检测纯度呢。这个化合物是通过1-boc-3-哌啶甲酸 和溴苄合成的, 还有1-boc-3-哌啶甲酸 的熔点是159-162℃。 我们这个东西的熔点是109-116℃。 所以这两个东西很定有是不一样的。 实在不行只有打核磁了。我们这个原料的紫外吸收也在204这个位置。但我们用254的紫外薄层检测,发现原料不显色,1-boc-3-苄基哌啶甲酸 轻微显色。[img=,281,247]https://ng1.17img.cn/bbsfiles/images/2019/07/201907231115067664_5937_1815404_3.jpg!w281x247.jpg[/img]
本人在岛津GC-2014上做样,FID检测器, 进样口:200,检测器:250,此样品以乙醇为溶剂,溶解以样品限度为浓度的吡啶0.004mg/ml 、哌啶0.004mg/ml、,在DB-624上做过,柱长:30m*0.32mm*1.80um, 进样:1.0ul 以40°C,8min,以20°每分钟升到200°C,保持3分钟,分流20:1、 10:1、5:1都测试过了,可哌啶就是不出峰,从浓溶液定位的图谱来看,哌啶和吡啶出峰时间相差0.1min,其实也就是难以分离,为此换hp-5柱子定位,可是哌啶和吡啶的峰总是拖尾,出峰时间虽然相差0.4min,但是实际按照以上的分流条件做样时候,哌啶不出峰,若是再修改分流比恐怕乙醇过载,杂质变大,更不好判断,于是又换了DM-1色谱柱,同样条件,浓溶液定位,这次哌啶在这柱子上根本不出峰,大浓度也不出峰,无奈又换了DM-WAX柱子做样,同样条件,同样定位,吡啶倒是峰形很好,可哌啶,居然引得基线上升,然后又持平,像是斜坡的样子,但是根本不积分!所以,本人想请问,各位兄台,各位高手,有没有一根柱子是对哌啶响应值高,且完美的分离吡啶和哌啶的,请指教!
本人要测定一样品中的哌啶含量,以哌啶纯品进样时出峰,以DMF做纯品的溶剂配成1mg/ml溶液时,哌啶不出峰,DMSO作溶剂时情况一样,但在大约1ml的DMF中加入两三滴纯哌啶时,哌啶出峰,而且峰面积不少,大概换算到1mg/ml的浓度也应该可以检出(1滴大概4~5mg),请问为什么会这样呢?大家有检过哌啶吗,用的是什么溶剂呢?我的检测条件是DB-624,进样口200度,分流比10:1,恒流2ml/min,程序升温40度(5min),15°C/min,220(5min),检测器FID250度,载气是氮气,请大家指教下!
有哪位朋友做过哌啶基哌啶的分析,能否跟我联系下,有一些问题想请教谢谢
[color=#444444]ESI 质谱条件下,M+H的二级产生含羰基的加氢峰,m/z 220,M+Na的二级碎片产生含羟基的加钠峰m/z 244,二者相差24。羰基氧和羟基氧与钠离子和氢离子的结合能力是怎样?[/color][color=#444444]难道钠离子更容易稳定含羟基离子?氢离子更容易稳定含羰基的离子?该怎么解释呢?[/color]
现在要做哌啶,N-甲酰基哌啶的含量,都是微量的,用了DB-624,DB-waxetr的测1000ppm的峰形都不好,请问有推荐色谱柱和方法么
2010年版药典氟哌啶醇片红外鉴别方法是:取本品粉末适量(相当氟哌啶醇约50mg),用20ml三氯甲烷分次研磨使溶解,过滤,取滤液水浴蒸干,残渣减压干燥后,压片,与标准图谱比较. 因为氟哌啶醇易溶于三氯甲烷,我在做的过程中就没有剥片;另氟哌啶醇对光 热具不稳定性,滤液采用60度水浴蒸至近干,采用药典原料项下的方法干燥:即60度减压干燥,约5小时,但取出压片时发现它的性状为半固体,压出的片透明度不好,还粘模具,没法做! 会不会是辅料有影响呢?想请教一下,如何改进我的试验,压制一个好的片子?
现在要做哌啶,N-甲酰基哌啶的含量,都是微量的,用了DB-624,DB-waxetr的测1000ppm的峰形都不好,请问有推荐色谱柱和方法么
有没有人检测过N-甲基哌啶
过氧化值的单位在新旧标准中有所不同,有g/100g,mmol/kg,旧标准中则是meq/kg而羰基价的单位只见有meq/kg ,未见其他现在不知道大家在平时检测过程中是如何使用这个单位的知识听说meq/kg不是正规单位,停止使用了这个在哪里有这方面具体的规定呀?
哌啶和乙醇

[color=#DC143C]五羰基铁[/color][img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911232034_186121_1610969_3.jpg[/img]Fe(CO)5为CO与Fe的合成物,化学反应方程为:5CO+Fe→Fe(CO)5。 [color=#00008B]Fe(CO)5的物理性质:[/color] 1、铁属于过渡元素,在它的原子中产生充填不满结构的电子层,在与一氧化碳相互作用下形成Fe(CO)5时,由铁原子与5个CO分子组成中获取不足的电子。其分子结构式如图: 2、在常压下,Fe(CO)5的熔点在-20.3℃左右,沸点在103.6℃左右,临界温度286℃左右。在100℃以下没有明显分解,100℃-130℃约有1%的分解140℃ -160℃有3.3%弱分解,160℃特别是179以上时,普遍强烈分解。 [color=#DC143C] Fe(CO)5的化学性质:[/color] 1、Fe(CO)5完全溶解于汽油、苯、四氯化萘、苯醛、丙酮、溴化苯、二氯化苯和其它溶液。 2、从-15℃起火花时,羰基物蒸汽与空气混合物一定产生燃烧,在温度34℃ 时(亦有报道60℃)就在适当条件下能够自燃。 3、Fe(CO)5相当的活泼,容易形成氢化羰基物H2Fe(CO)4及其金属盐Na2Fe(CO)4,卤化羰基物Fe(CO)4I2、亚硝酰基羰基物Fe(CO)2(NO)2、氯化羰基物Fe(CO)3(CH3OH)、环戊二烯羰基物[C5H5Fe(CO)2]2等很多化合物。 4、Fe(CO)5光化学性能很好,在光的作用下Fe(CO)5分解形成Fe2(CO)9。 5、当加热到140℃时,Fe(CO)5易氧化,形成Fe2O3(铁氧体)。 6、针对Fe(CO)5的临界温度,在常压及温度在250℃-300℃时进行Fe(CO)5的热解,是Fe(CO)5最重要的应用,是工业化制取羰基铁粉的最基本方法。
各位[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]界的达人,求助各位一个问题: 有个氨基哌啶,成品盐酸盐,无紫外吸收,但也不想采用滴定的方法分析,那么,采用哪种比较适宜的GC固定性和条件分析这个氨基哌啶盐酸盐的含量呢?是否采用固定相为SE-54 或PEG20000的这两种不同极性的都可以呢?可检索到的关于哌啶或者哌啶盐酸盐的分析方法太少了啊。在此积极求助,忘各位指导迷津啊。
样气里面含有硫化氢和羰基硫,但标气里面只有羰基硫,怎么标定色谱啊?
求教用顶空做哌啶和DMF溶剂残留的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件设置,目前试过DB-624 ,HP-5柱子,db-624可以让两试剂出峰,但是哌啶出峰情况不稳定且很容易残留,峰面积时大时小,且混合样中甚至可能不出峰,DMF出峰情况很稳定,但是峰面积过小。试过提高顶空平衡温度,效果不佳。HP-5,DMF不出峰,哌啶出峰情况较DB-624好,峰面积稳定,拖尾不严重。
大家好! 我有一个正壬烷试剂样品,用GB/T6324.5-2008测定羰基数高于100ppm,但是,该样品送质谱,色质联用、OFID检测,均为发现含氧化合物,请高手帮忙分析,非常感谢!
我有个3-羟基哌啶盐酸盐的样品,用GC检测,先将盐酸盐处理掉后,成3-羟基哌啶进气相色谱,FID检测器,色谱柱是DB-5和DB-17,峰形很难看,不规则的包,求助:用什么色谱柱会比较好?
1-BOC-4-哌啶甲酸检测其纯度时DB-624和HP-5严重拖尾,在DB-WAX中无法出峰,请问选择什么色谱柱合适?或有其它的检测方法
4-羟基哌啶用Aglient1260检测,流动相乙腈,甲醇,各种比例都试过,C18的长柱,短柱都用过,怎么都是不出主峰,求助给些线索!
我现在在合成双哌啶醇酯,反应之后无法分离形成的单酯和双酯,想通过滴定来判断单双酯的含量,请问各位大侠有没有什么好的方法!即测定六元环上的仲氨.
4-(4-羟基苯基)哌啶的液相条件,谢谢!
4-羟基哌啶溶于甲醇后,只有甲醇峰,样品再浓都没有?跟柱子有关系吗?
求问哪位大神用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS法检测过1-乙酰哌啶?具体仪器条件色谱柱流动相啥的,求提供参考。我用的是AB家的质谱4500
有哪位高人知道 ov101(30m)色谱柱能否分开哌啶和吡啶? 急,谢谢
最近要测羰基指数,样品为PTMEG(聚四亚甲基醚二醇)是是四氢呋喃的聚合物。测它的羰基指数怎么弄?一般找那个峰为基准?现在只知道有文献上写羰基指数CI=Ac=o/Aref..... 求高人指点
大家好!我有一个正壬烷样品,用GB/T6324.5-2008测定羰基数100ppm以上,但用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]、OFID 、MS都未检测到微量含氧化合物,请大家帮忙分析下是什么原因?
跟据药典方法设置仪器参数,使用了DB-624的柱子哌啶出峰不稳定,且有时不出峰,更换为DB-1301色谱柱哌啶也没有出峰,请教一下各位大神,应该如何解决这个问题呀
我们在测定甲醇中羰基化合物时,严格按照操作步骤进行,先制备无羰基甲醇,再按步骤一步一步进行,但当进行到加入氢氧化钠甲醇溶液时,过一会出现白色沉淀,如加入盐酸,沉淀又消失,为什么?
羰基a-H应该算活泼氢吧(因为可能形成烯醇结构),如果将含有羰基a-H的化合物溶于氘代甲醇,羰基a-H的共振峰是不是会消失(因为被氘代甲醇的羟基氘取代)?另外羰基a-H的化学位移是不是也像其他羟基氢一样处于低场,在0-10ppm的核磁共振图谱中一般不会显示?请高手指点,谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP