三氟化硼乙腈络合物为什么会变色
三价铁离子与什么会形成无色的络合物?
本人现在需要做一个稀土金属金属的检测,找到一个方法是显色剂与其络合,用HPLC检测该络合物。现在初步尝试了下,发现用乙腈和甲醇与缓冲液就算改变比例也没有保留,想看看有没有谁有相关经验做过金属络合物用HPLC检测的?接下来的想法是加入离子对试剂或者换亲水色谱柱,这些只是新手个人猜想,希望得到指正!

[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911121025_183976_1610969_3.jpg[/img][color=#DC143C]络合物 complex compound [/color] 配位化合物的旧称。按英文名称,络合物有两种含义:一种是指分子中含有配位键的化合物 另一种是不含配位键,而由特有的相互反应形成的聚集体,例如淀粉与碘形成的蓝色物质,抗原与抗体分子的结合物等。前一种化合物按照1980年中国化学会《无机化学命名原则》应称“配位化合物”。后一种宜称复杂化合物,简称复合物。 络合物之一 络合物通常指含有络离子的化合物,例如络盐[Ag(NH3)2]Cl、络酸H2[PtCl6]、络碱[Cu(NH3)4](OH)2等;也指不带电荷的络合分子,例如[Fe(SCN)3]、[Co(NH3)3Cl3]等。配合物又称络合物。 络合物的组成以[Cu(NH3)4]SO4为例说明如下: (1)络合物的形成体,常见的是过渡元素的阳离子,如Fe3+、Fe2+、Cu2+、Ag+、Pt2+等。 (2)配位体可以是分子,如NH3、H2O等,也可以是阴离子,如CN-、SCN-、F-、Cl-等。 (3)配位数是直接同中心离子(或原子)络合的配位体的数目,最常见的配位数是6和4。 络离子是由中心离子同配位体以配位键结合而成的,是具有一定稳定性的复杂离子。在形成配位键时,中心离子提供空轨道,配位体提供孤对电子。 络离子比较稳定,但在水溶液中也存在着电离平衡[1],例如: [Cu(NH3)4]2+=Cu2++4NH3 因此在[Cu(NH3)4]SO4溶液中,通入H2S时,容易生成CuS(极难溶)
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
工业盐中常含有分散剂铁氰络合物,但在化盐时常常要去除其中的铁离子,有谁知道其方法,或者是这种络合物的特性?以及它的测定方法?如果时用ICP测定,样品应该怎样处理?谢谢
有一种有机金属络合物甲基环戊二烯三羰基锰(MMT)含量分析,用什么方法检测?能否用GC?若用GC什么柱子最好?
问题描述:测定某金属络合有机物的样品,采用ESI直接进样测定可以同时得到有机物和金属有机络合物的质谱峰,但采用UPLC+ESI希望分离 测定却只检测到该有机物质谱峰。流动相是水(醋酸铵)+乙腈,色谱柱为UPLC-C18。请教各位,哪个环节可能出了问题?感谢
金属络合物可以做核磁吗?做出来的谱图不是很理想。
有谁知道EDTA-铜铵络合物的质量指标,脆求!谢谢!
络合物组成的测试方法【作 者】薛光等编著 【形态项】 228 ; 20cm 【出版项】 成都市:四川科学技术出版社 , 1985 【中图法分类号】O641.4
硫酸亚铁铵将黄色的硅钼酸盐络合物还原成兰色的硅钼兰络合物,然后光电分析法开始,我是小白,我想知道为什么用硅钼蓝络合物?哪位大神告诉下,在线等
主要是想测定消解后的效果,故想自己做一种有机铜络合物来高温酸化消解一下来看下消解的效果。有哪位大侠知道的给一些建议。谢谢,有资料的也可以给我发邮件:slivyer@yahoo.com.cn,
【题名】:以多元混配络合物为电活性物的设计思想及初步实验【全文链接】: https://www.cnki.com.cn/Article/CJFDTOTAL-HXCH198801002.htm
如题,请教各位如何用气相色谱测试“铂-四甲基二乙烯基二硅氧烷”络合物的铂含量?有没有标准分析方法?望大家不吝赐教!谢谢!
【题名】:中性载体络合物在离子选择性电极中的应用【全文链接】: https://www.cnki.com.cn/Article/CJFDTOTAL-HXTB198002001.htm
我的八羟基喹啉的金属络合物的NMR能不能测?是不是如果仅与金属离子的顺磁性有关?顺磁性是不是仅影响C谱?H谱无关?
铝试剂与金属离子形成的络合物带正电还是带负电??如果和铜离子络合,带什么电,带几价电荷?请长辈多多指点,感激不尽。
[color=#444444]本人现要确定反应液中催化剂(分子量一千以上金属络合物)的残余量,该催化剂跑板在紫外条件下显色,但是使用多种展开剂均不移动,液相色谱也不出峰(使用C-18柱),请各位前辈指教!有什么方法能对它进行定量分析?[/color]
[color=#444444]三乙胺-丙酮-盐的混合液,三乙胺的含量在5%以下,请问有什么好的方法可以测定三乙胺的含量,在实验室用氢火焰毛细柱打可以出峰,但是如果含量特别低时担心[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]打不出,导致微量三乙胺检测不出来。查文献有溴酚蓝分光光度法、红外色谱,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]、电位返滴定法,以及混合液放入盐酸,用过量NaOH滴定未反应酸,溴酚蓝做指示剂等方法。请问这几种方法哪个比较准确,并且好操作,谢谢各位了![/color]
EDTA和锌的络合物在水中还能与NaOH反应生成Zn(OH)2沉淀吗?络合物和碱反应是怎么反应的?
当溶液中的二价铁离子是络合物的状态存在,再向溶液中加入氢氧化钠,是否会生成[font=arial, 宋体, sans-serif][color=#333333]氢氧化亚铁沉淀[/color][/font]
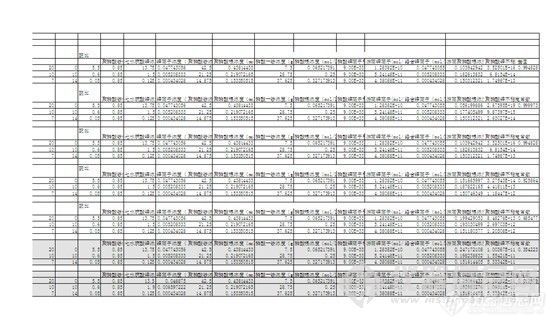
进行这项计算的最根本的目的是,希望通过理论计算,指导生产实际中微量元素的添加量,避免每个配方都重复进行实验。进一步的,是在聚磷酸铵、磷酸一铵的比例为给定值时,添加最大量的七水硫酸锌,而不致产生沉淀。计算过程中遇到的大问题有如下几个 :一,理论基础差,直到现在,我还不敢保证所有计算中使用的关于络合物的理论是完全正确的;二,聚磷酸铵是一种混合物,我们使用的是水溶性聚磷酸铵,由二聚体、三聚体、四聚体……组成,n(聚合度)最高不超过20,论坛里初晶古恒老师指出,不同聚合度的聚磷酸铵,其络合能力是不一样的,我在处理中,忽略了这一因素,而是把聚磷酸铵当成某一分子量的“纯物质”,不知道这个近似对最终结果的影响有多大;三,聚磷酸铵-锌络合物的组成不确定,我在论坛里的上一个帖子http://bbs.instrument.com.cn/shtml/20120716/4144753/ 中,认为聚磷酸铵与锌离子是1:1反应,但是在后面的实验和计算中,我又推翻了这个假设。在本文里我对聚磷酸铵-锌络合物的配位数进行了探讨,尚且不敢打包票说是正确的。我参考了马娟等的“水溶性聚磷酸铵制备微量元素螯合物的实验研究”,对聚磷酸铵-锌络合物的配合物有一定的指导,但是不能彻底解决上述问题。背着这“三座大山”,我再度开始了实验与计算。我的思路如下:根据各物质的添加量,计算出溶液中“聚磷酸根”、磷酸根(认为磷酸一铵完全解离)、锌离子的表观浓度(mol/L)(不知道表观浓度这个词语用的对不对啊),认为溶液中的磷酸根全部是游离的,根据磷酸锌的离子积(9.0*10^-33),计算出溶液中游离锌离子的浓度A,全部的锌离子减去游离的锌离子,就是参与络合反应的锌离子B;再根据锌离子与聚磷酸铵络合的配位数(下文详述),计算出参与络合反应的聚磷酸根的量C,总量扣除参与络合反应的聚磷酸根的量,即为游离聚磷酸根的量D;据此计算出聚磷酸合锌的不稳定常数。不同实验数据,得出的聚磷酸合锌的不稳定常数,应该是一致的。实验数据共有四组,考虑到锌的添加量很小时,相对误差较大,因此只要前两组数据计得的不稳定常数一致,即认为方法成立。不稳定常数的计算公式:K=2+]*[PP][sup]n/[ZnPP],式中2+]为体系中游离锌离子的浓度A,[PP]为游离聚磷酸根的浓度(按单体算)D,n为锌与聚磷酸根络合反应的配位数,[ZnPP]为聚磷酸锌的浓度B。实验中使用的聚磷酸铵纯度为85%,另外15%为磷酸一铵。对于前两组实验数据,设n分别为8、7、6、5、4,计算聚磷酸锌的不稳定常数,发现当n为4时,计得的不稳定常数值最为接近。再对实验数据进行微调,可使得到的不稳定常数一致。又据查阅文献,一般没有三配位数的络合物,锌也几乎不存在二配位数的可能,因此认为该络合物的配位数为四。至此,络合物组成和不稳定常数都得出了结论。并根据得到的数据,预测了聚磷酸铵为15g,磷酸一铵为5g及聚磷酸铵为14g,磷酸一铵为7g时,应添加的七水硫酸锌的量,经实验,实验结果与计算值较接近。另外,较通用的络合物组成的实验方法为分光光度法,本实验还需用分光光度法进行验证。所有实验数据及计算过程见下图。实验中所有组合的溶解方法均为20g磷肥加400毫升水。
顶空[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测三乙胺,谁有相关经验吗?设置条件大概是多少?三乙胺溶液里面需要添加碱溶液吗?求回复
最近在做水质三乙胺的检测,发现校准曲线很难做直,请问主要有哪些影响因素,还有三乙胺的标样何处可以买到?
求助铁钴镍金属氧化物及他们的有机络合物的拉曼特征峰位置,谢谢
各位老师。我最近在做一个化合的异构体分离,流动相中需要加如三乙胺扫尾,但是发现采用三乙胺扫尾后,色谱峰比不加三乙胺的保留时间要延迟很多,这是什么原因呢?我一直认为三乙胺是不会影响手性保留的!大家有什么高见?
刚接触分析没多长时间,公司领导要求我做一个车间回收三乙胺定量的方法,面积百分比不准,然后就尝试做一个外标法,因为试样当中有邻二氯苯和三乙胺两种物质,所以我就配置了不同浓度的三乙胺溶液,5%.10%.15%.20%.25%.的溶液,用自动进样器进样,发现峰面积相差很大,这是为什么?到底该怎么做呢?求助前辈~我的气谱方法是这样的,进样0.2ul,分流比100:1,进样口气化温度250,检测器300,柱箱150保持5分钟。HP-5中性柱,安捷伦7820A,FID检测器
请问用什么填料的色谱柱分离叶酸和叶酸络合物较好?
小弟分析生物碱,用的流动相为甲醇/水(含0.14%H3PO4和0.5%三乙胺),防拖尾效果不错。 但0.5%三乙胺是不是太高了?会对柱子不好吗?望论坛高手释疑







 我要推广仪器
我要推广仪器
 下载APP
下载APP