请问哪位版友有农产品中三氯异氰尿酸、三乙膦酸铝、氟苯脲、氟吡甲禾灵、氟酰胺、环酰菌胺的测定方法? 三氯异氰尿酸在棉花、水稻上的测定方法,三乙膦酸铝在蔬菜、水果中的测定方法,氟苯脲在蔬菜、水果中的测定方法,氟吡甲禾灵在水果、咖啡豆中的测定方法,氟酰胺在稻米中测定方法,环酰菌胺在蔬菜、水果及其干制品中的测定方法。最好是国标或行标。先致谢了。
有这么一种流动相,甲醇:水:三乙胺:乙酸=600:400:1:1那么加三乙胺和乙酸的作用是什么?
行标YC 144-2008中规定三乙酸甘油酯纯度测定时,气相进样口温度为250℃。但我查到的三乙酸甘油酯沸点为:258-260 °C(lit.)℃。进样口的温度设置原则不应该是要高于被分析物的沸点,确保所有分析物经过进样口进样后能够完全气化吗?想寻求大家的解答
安谱三乙酸甘油酯的使用心得 我们实验室一直在测烟标中的VOC,参照烟标YC/T 207-2014的方法来检测,YC/T207-2014同YC/T207-2006相比培加了10种管控物质,同时对溶剂杂质也有要求,基于方法的加严,为了达到更好的检测数据,对检测用的试剂自然而然的加严了,所以,就采购的安谱药典级的三乙酸甘油酯。以前我们使用的某品牌的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819191794_01_2769262_3.jpg现在我们使用的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819194869_01_2769262_3.jpg现就两种不同的三乙酸甘油酯,做个对比测试,选择用新的20ml顶空进样瓶,然后分别移取不同品牌的三乙酸甘油酯到顶空进样瓶中,封盖,然后用顶空加气相(带FID检测器)检测,检测结果如下:某品牌的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223603_01_2769262_3.jpg安谱药典级的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223175_01_2769262_3.jpg两种品牌试剂的检测结果汇总表:http://ng1.17img.cn/bbsfiles/images/2015/09/201509181923_566740_2769262_3.jpg综上,安谱药典级的三乙酸甘油酯优于某品牌的三乙酸甘油酯,安谱药典级的三乙酸甘油酯值得版友们拥有。
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
RT,做三氯化磷,三氯化氧磷,五氯化磷,三氯硫磷,磷酸三乙酯的朋友有没?本人QQ444980036大家一起聊聊啊,最好是质检方面的,嘿嘿!!
最近在尝试开发REACH的一些检测方法,很有难度啊。其中三乙基砷酸酯怎么测试,请大家帮忙一下,不胜感激![~113806~]
三乙基砷酸酯是无机物吗?
通常液相色谱分析中,在流动相中加乙酸和三乙胺,大家来谈谈,在什么情况下需要加乙酸,什么情况下加三乙胺,加多少是怎么控制的?
原子吸收测定三乙酸甘油酯、沒食子酸丙酯中铅、镉的含量?那位做过,我现在查的沒食子酸丙酯中铅的测定按照GB/T5009.75测定,请问哪里有标准下载。三乙酸甘油酯、沒食子酸丙酯中铅镉的含量都是微量
原丙酸三乙酯大浓度定位也还无法确定在哪出峰,目前是peg20的柱子 溶剂还没找到太合适的
原子荧光测定三乙酸甘油酯、沒食子酸丙酯中砷、汞的含量?含量是微量的,样品是采用湿法消解还是采用微波消解?
在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中三乙胺与醋酸异丙酯怎样有效地分离?
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先 补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论跟上次的问题一样,上次结贴太匆忙了,再次征集。感谢上次renture/jiangyunjun3/melu的帮助,找到一个分光法测痕量氮川的方法,测量范围是0.01~0.12ug/mL,但我们的样品是含量大概有0~10%左右的氮川,是否能够通过稀释来使用上面的分光方法还不知道。现在想征集一个能够分析常量氮川的方法,希望大家踊跃发言。
三乙酸甘油酯+硝酸,放在电热板上100度加热,会有危险吗?可能的反应产物是什么,谢谢大家
怎么分离亚氨基二乙酸和氨基三乙酸,样品中氨基三乙酸含量少
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先[em0813] [em0813] [em0813]补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论
2008年1月18日,美国EPA宣布建立了以下杀虫剂的注册审议文档。随本文件,EPA开始相关注册审议的公众评议期。注册审议是EPA为保证每种杀虫剂始终符合注册法规标准,意即:杀虫剂在对人类健康或环境没有过分负面影响的条件下起到其预定的作用,而对注册杀虫剂定期进行的审议。注册审议文案包括有助于公众了解EPA在注册审议过程中要考虑的信息及问题。 通过该计划,EPA可保证每种杀虫剂注册都能以当前科学及其它知识为依据,包括对人类健康和环境的影响。三乙膦酸铝(Fosetyl-Al (Aliette) EPA-HQ-OPP-2007-03单氰胺Cyanamide EPA-HQ-OPP-2007-10硫代碳酸钠Sodium Tetrathiocarbonate EPA-HQ-OPP-2007-10喹禾灵Quizalofop EPA-HQ-OPP-2007-10恶草平Isoxaben EPA-HQ-OPP-2007-10二氯喹啉酸Quinchlorac EPA-HQ-OPP-2007-11氟虫胺Sulfluramid EPA-HQ-OPP-2007-10 该通知以后是否会采取进一步措施尚不能定。 G/SPS/N/USA/1751
请问EDTA中氨基三乙酸的含量,不合格的批次多吗?
标题:出处:安徽大学学报:自然科学版 1999年23卷3期作者:聂康明[1] 李蕤[2] [1]安徽大学化学系 [2]合肥联合大学化工系 摘要:以甲基丙烯酸和二缩三乙二醇为原料,合成用于制备PP/PnBA IPN共混材料的交联剂--双内烯酸二缩三乙二醇酯,应用傅里叶红谚氢核磁共振谱(H-NMR)表征了产物的化学结构,分析表明,填充互穿聚合物组合组分的相容性交联剂的化学结构有关,长链柔顺性罗好的双人烯酸二缩三乙二醇酯使填充IPN共混体系双组分的Tg值内移程度较大,表现出较好的强迫增容效果。
[color=#444444]各位大神给点建议,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]所用的流动相是三氟乙酸和三乙胺配制的缓冲液,来检测发酵液中的有机酸,能不能给点建议,具体怎样配制此缓冲液?[/color]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=126764]Triethyl arsenate三乙基砷酸酯[/url]这是其中Triethyl arsenate三乙基砷酸酯的性质!后续还有怎么评估物品种是否含有!
我手工过柱子,分离约15克的含N化合物,点板发现拖尾严重,流动相加入三乙胺或者氨水点板,就收到明显效果,不再拖尾。所以过柱子的时候,石油醚-乙酸乙酯流动相中需要加入氨水或者三乙胺。但是,加入氨水的话,氨水含有很多水,跟石油醚等互不相溶;加入三乙胺,师兄说很难蒸发除掉,会混在我的产物里面。因此,求助于各位高手,我过柱子的时候,到底该加什么?
GC,我用色谱柱TG-624 检测三乙胺、乙酸异丙酯和庚烷的混合样,分离总是不好。该如何调节?
今天用液相色谱仪检测依地酸钙钠中的氨基三乙酸,用的是安捷伦的C8柱子,流动相为0.01mol/L的氢氧化四丁基铵:甲醇=90:10,溶剂为硝酸铜,可是压力很不稳定,好不容易平衡后,一进样,压力瞬间飘很高,在走样过程中逐渐下降,走一针过后,基线很不稳定,感觉柱子里面冲出来很多东西,有没有做过这个品种的前辈们啊,知道我一下吧,怎么改进?
有网友问三乙醇胺哪个厂家好?他在用紫外法做水中砷,发现用三乙醇胺配制的吸收液空白吸光度值高,大家都用什么厂家的三乙醇胺?
做氨基酸用的药品三乙胺,醋酸钠方法上说需要优级纯,可是优级纯的很难买到,用分析纯的可以不?
大家好,有谁知道磷酸三乙酯的提纯方法
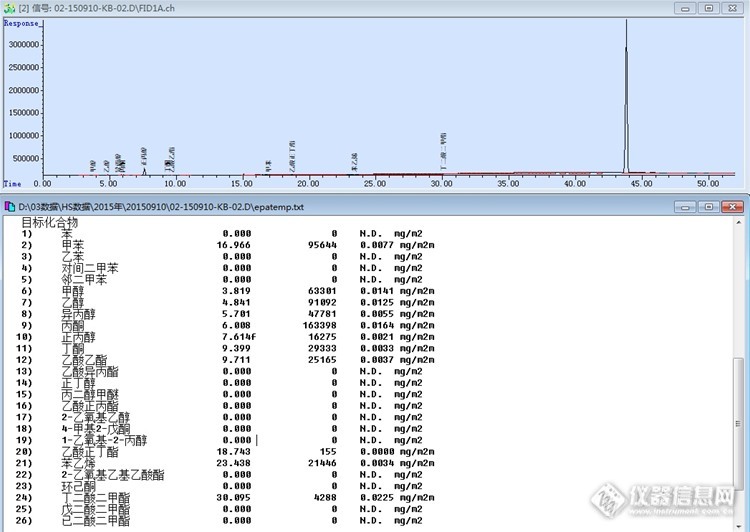
各位老师: 大家好,我们公司在用气相检测磷酸三乙酯的含量,但是遇到了一点问题,不知从何下手解决,所以请各位老师帮忙分析一下我附的谱图里面都是什么物质,第一个峰是空气,第二个峰是水,然后没分开的小峰是乙醇,8分半出的是三乙酯,其余就不知道了,正常情况是8-9分钟出三乙酯的峰,但是1.8min出了一个大峰,虽然这个是没有反应完全的三乙酯,我怀疑是HCL的峰,但是工程师说了不可以出HCL的峰,会影响柱子的寿命,所以,我该怎么去验证这些峰是什么东西啊,谢谢了! 仪器型号:agilent GC 7890A 色谱柱:HP-5(30m ×0.530mm ,5.00μm ,60-260℃) 检测器:TCD 载气:氦气 方法:进样量:1μL 升温程序: 70℃ 保持 2分钟 20℃/min 升到200℃ 保持4min 分流比: 10:1,60Ml/min 进样口温度:260℃
有没有用到在流动相中加入三乙胺,再用盐酸调Ph的呢







 我要推广仪器
我要推广仪器
 下载APP
下载APP