(2S,3S)-3-氨基-2-甲基-4-氧代氮杂环丁烷-1-磺酸(2S,3S)-3-amino-2-methyl-4-oxoazetidine-1-sulfonic acid谁有这个液相的检测纯度的方法,是氨曲南的起始物料N-氮杂环丁烷???
[size=2]环境保护部公布的“HJ504-2009 [color=#cc0000]环境空气臭氧的测定靛蓝二磺酸钠分光光度法”,[/color][color=#000000]是对原国家环境保护局1995年3月25日批准、发布的《环境空气臭氧的测定靛蓝二磺酸钠分光光度法》的修订。此标准适用于环境空气中臭氧的测定,相对封闭环境(例如室内、车内等),空气中臭氧的测定也可参照本标准。我国室内空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量标准规定,空气中的臭氧浓度不应大于0.16毫克/立方米,如果人所处环境高于这个标准,时间长了,就会产生不良反应。新修订的该测量标准,修改了标准和适用范围,增加了测定上限和测定下限。具体的方法原理:空气中的臭氧在磷酸盐缓冲剂存在下,与吸收液中的蓝色靛蓝二磺酸钠等摩尔反应,褪色生成靛红二磺酸钠,在610nm处测量吸光度。标准原文:[url]http://bbs.instrument.com.cn/shtml/20091109/2201749/[/url][/color][/size]
想请教十水硫酸钠含量测定(杂质有:硼酸钠\对甲苯磺酸),我想用钡盐沉淀重量法,但怕杂质干扰,也想用强阳离子树脂交换法将钠交换出来后用氢氧化钠滴定,但也怕杂质干扰,不知还有什么方法检测,请大家不怜赐教!
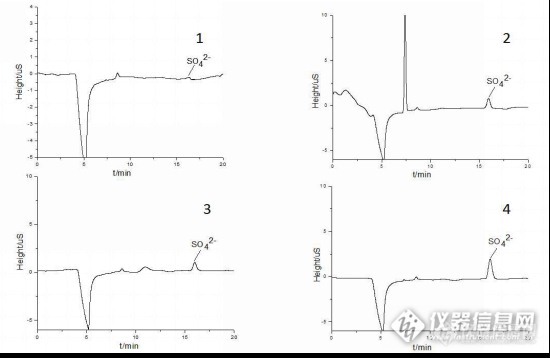
在线固相萃取-离子色谱法测定四种芳环磺酸盐中的硫酸根离子摘要:建立一种在线固相萃取离子色谱法测定四种芳环磺酸盐中硫酸根离子含量的新方法,将自装填的PGC-SPE柱应用于离子色谱系统对样品进行在线前处理,样品经过PGC-SPE柱处理后进入500μL定量环,通过阀切换-大体积进样模式使硫酸根进入阴离子检测系统。固相萃取流路以1.5mmol/L Na2CO3在0.8mL/min的流速对基体在线富集;分析柱采用SH-AC-3 (4.0×250mm) + SH-AG-3 (4.0×50mm),在6mmol/L Na2CO3 +4mmol/L NaHCO3条件下等度洗脱,柱温为35 ℃,流速为0.8mL/min,进样量20μL。结果表明:硫酸根离子在0.50-10.00 mg/L浓度范围内呈良好的线性关系,线性相关系数为0.9993,保留时间、峰高、峰面积的RSD均在0.28%~2.86%之间,方法检出限为0.0106mg/L,回收率在93.33%~105.59%之间;该方法具有良好的线性和重复性,整个在线分析过程在25min之内完成,进样量少,快速、高效。关键字:在线固相萃取,离子色谱,多孔石墨化碳,自装填技术,硫酸根离子1 前言多孔石墨化碳(PGC)作为一种新型色谱固定相,因其表面为纯碳结构、平整且无接枝的功能基团而具有耐强酸强碱、耐高温、性能稳定的特点,广泛应用于液相色谱和离子色谱中。PGC是二维结构、表面完全非极性且有可自由移动的π电子,因此与它化合物之间存在着疏水作用和电子间作用等多重作用力,从而在一定条件下对极性化合物、非极性化合物、异构体、聚合物等都具有保留性且对强极性化合物表现出强保留性。在样品前处理特别是复杂样品前处理过程中,通常利用固相萃取对样品进行净化与浓缩富集,除去干扰性的杂质,从而提高待测物分离度并保护色谱柱。Thermo公司首先推出以多孔石墨化碳填料为固定相的hypercarb 分析柱。在固相萃取技术应用中已有商品化的离线多孔石墨化碳固相萃取小柱(PGC-SPE柱),主要应用于液相色谱和离子色谱的样品的富集和基体消除。 萘磺酸盐和蒽醌磺酸盐属于多苯环磺酸盐,易溶于水的强极性离子型化合物。目前商品化的离子色谱固相萃取前处理柱,如:Onguard RP柱、H柱、Na柱以及聚合物树脂固相萃取柱等对非离子型有机化合物和常规离子基质有很好的前处理效果,但对芳环磺酸盐类的离子型强极性有机化合物保留性差,前处理效果不理想;在测定这类物质中残留的无机离子时,若不能很好地对该类物质进行基体消除,其进入分析系统后易保留在离子色谱柱上且较难洗脱,对分析柱造成损害。PGC因其特殊的表面疏水性质和带电性质使得它对该类强极性离子型化合物具有很强的保留性。本实验创新性的提出自装填可再生的多孔石墨化碳固相萃取柱并将其应用于在线离子色谱分析系统对四种芳环磺酸盐中的硫酸根离子进行测定,自装填和可再生重复使用的特点很大程度上降低了实验成本,在线基体消除较离线前处理在能保证理想的前处理效果和样品分析重复性的同时,能很大程度的减少样品污染并缩短了分析时间和样品量,更有利于复杂样品中阴离子的快速、准确的分离分析。2 实验部分1.1 仪器与试剂实验所用仪器为自组装离子色谱系统,主要部件包括:Ultimate3000 WPS-3000TSL自动进样器,ICS5000+紫外检测器,ICS5000+(SP/DP)双泵,ICS5000 TCC柱温箱(含一个六通阀和一个十通阀),ICS3000(SP)单泵,ED50A电化学检测器(DS3,带控温)Chromeleon6.8色谱工作站,(美国赛默飞世尔科技有限公司);WLK-6A阴离子抑制器(青岛仪趣仪器有限公司);AL-10电子天平(梅特勒-托利多有限公司);Milli-Q Advantage A10超纯水机(Millipore);BRANSON 2510超声清洗仪(BRANSON); 硫酸根离子标准储备液(100 mg/L,上海计量测试技术研究院);碳酸钠和碳酸氢钠(分析纯,99%,上海凌峰化学试剂有限公司);氢氧化钠 (w/w=50% ,默克公司);浓磷酸(分析纯,国药集团化学试剂有限公司);甲基磺酸(99.5%,上海阿拉丁有限公司);甲醇,乙腈(色谱纯,上海星可高纯溶剂有限公司)。 1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾(上海翰思化工有限公司提供)1.2 标准溶液的配制 分别取0.50,1.00,2.00,5.00,10.00 mL的SO42-标准储备液于100 mL容量瓶,超纯水定容后得到0.50,1.00,2.00,5.00,10.00 mg/L的SO42-标准液。 准确称取0.0242g、0.0213g、0.0248g、0.0257g的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾于50mL容量瓶,超纯水溶解定容,作为待测样品。1.3 色谱条件在线PGC-SPE柱为本实验室自装填前处理小柱(3.0×30mm),PGC填料粒径:30μm,阴离子分析柱: SH-AC-3 (4.0×250mm,9μm) + SH-AG-3 (4.0×50mm,12μm) (青岛盛翰色谱技术有限公司),柱温:35 ℃,流速:0.8mL/min;进样量20μL;定量环:500μL;淋洗液:6mmol/LNa2CO3 +4mmol/LNaHCO3; 分析时间25min;1.4 On-line SPE-IC系统的构建 On-line PGC-SPE-IC系统构建如图1 所示,实验建立了阀切换-大体积进样抑制电导离子色谱方法测定四种芳环磺酸盐中的硫酸根离子。系统中泵1用于在线固相萃取和进样,泵2用于阴离子分析,泵3用于SPE柱的在线清洗再生;利用一个十通阀和一个六通阀实现样品的在线固相萃取、大体积进样和在线前处理柱的再生,抑制电导检测器对硫酸根离子进行检测。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131609_609526_3137073_3.jpg 图1 在线SPE-IC系统2 结果与讨论2.1 PGC-SPE柱的装填和活化及柱容量 实验中所使用的SPE柱为本实验室自装填前处理小柱,采用湿法装填方法,以乙醇为分散剂,将填料以流动相的形式利用高压泵进行装填,装填过程先是填料在低流速低压条件下缓慢泵入柱体内,一段时间后再缓慢将流速调节为1mL/min装填1h以上,最后加大流速在高压条件下将填料压实;装填好的SPE柱压力约为120 psi。将装填好的PGC-SPE柱进行一下酸碱活化过程:酸冲洗(100mmol/LHCl, 1mL/min,30min)——水冲洗(1mL/min,20min)——碱冲洗(100mmol/LNaOH, 1mL/min,30min)) —— 水洗(1mL/min,20min)。酸碱冲洗的作用在于对小柱进行活化同时溶解填料中本身残留的一些酸碱可溶性杂质,避免其进入色谱系统损害色谱柱和抑制器。 利用紫外检测基线突越的方法测定PGC-SPE柱对该四种化合物的柱容量。分别配置500mL浓度为230.0、236.4、129.0、258.0mg/L的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾溶液作为流动相,以0.1mL/min的流速流过SPE柱,记录基线开始采集和发生突越的时间间隔,根据公式计算(柱容量=物质浓度*流速*时间)得:1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾的柱容量分别是0.8912mg,0.8307mg,0.2354mg和0.4373mg。2.2 色谱分析条件的选择2.2.1 硫酸根在SPE柱上洗脱条件(泵1淋洗液) 在中性条件下, 由于电荷的相互作用,PGC-SPE柱对硫酸根具一定的保留性,而酸性或者碱性条件下将其洗脱;实验中考虑到仪器系统兼容性和耐碱程度,选择低浓度碳酸钠作为洗脱液。由于使用500uL定量环大体积进样方式,流速为0.8mL/min,则SO42-的峰展宽必须控制在0.625min之内。根据实验结果可知(见图2),在0.5mM,1mM,1.5mM碳酸钠条件下,阴离子完全洗脱时间分别是1.10-2.20min,0.75-1.45min,0.65-1.20min;1.5mM浓度下的硫酸根离子在0.55min之内洗脱,因此Pump1选择1.5mM碳酸钠作为洗脱液。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131601_609519_3137073_3.jpg 图
GCMS测定磺酸类基因毒杂质如何选择定性离子,定量离子?,已知磺酸类基因毒杂质为三氟甲磺酸乙酯,GCMS 法,SIM条件的质谱条件如何确定?
做铁的时候,二苯胺磺酸钠对氧化还原滴定的影响程度有多大?
最近强生婴儿用品有毒事件很火,据检测报告显示甲醛和1,4-二氧杂环乙烷导致过敏,哪位大虾可以提供关于1,4-二氧杂环乙烷的信息?包括CAS号,结构式等,谢谢!E-mail: ljmw521@163.com
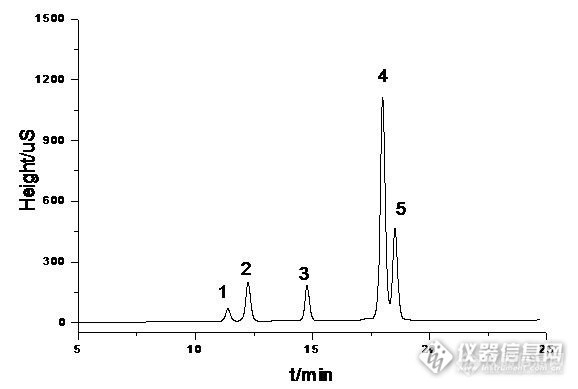
五种芳环磺酸盐的液相色谱分离方法浅谈 1-萘磺酸钠,2-萘磺酸钠,蒽醌1,5-二磺酸钠、蒽醌1,8-二磺酸钾和氨基萘酚-二磺酸钠五种物质均为强极性芳环磺酸盐化合物,在水溶液中完全电离成离子状态。在液相色谱分析中,五种物质的保留性质相似;因其为离子状态,C18反相柱对其没有保留,无法将其成功分离。本分析方法中采用离子对-反相色谱方法对其进行分离。具体分析条件和色谱分离图如下: 表1 色谱分析条件分析仪器U3000 HPLC 系统 色谱柱Diamonsil®(钻石)C18200*4.6mm,5um流动相A乙腈梯度洗脱:30minA: 20% ___ 45%B0.1%TBA+0.3%磷酸二氢钾pH6.5 温度30℃ 流速1mL/min 检测波长222nm http://ng1.17img.cn/bbsfiles/images/2016/09/201609141634_609777_3137073_3.jpg 图1 五种芳环磺酸盐的液相色谱分离图 1:蒽醌1,5-二磺酸钠,2:氨基萘酚-二磺酸钠,3:蒽醌1,8-二磺酸钾,4:1-萘磺酸钠,5:2-萘磺酸钠总结: 从色谱条件分析,本方法使用的是常规的反相C18色谱柱,在流动相中加入离子对试剂的方法增强保留性,用磷酸二氢钾调节pH至近中性。方法建立过程中发现pH和乙腈的比例对五种物质的分离影响较大,溶液的pH的大小影响芳环磺酸盐在水溶液中的分子形态,酸性溶液中,各物质形成成分子,中性和碱性条件下电离成离子状态,碱性溶液中有利于与离子对试剂的结合,但考虑到仪器和色谱柱不耐碱性能,选择中性范围较为合适。流动相中乙腈的比例对各物质在色谱柱中的保留时间影响。经过多次试验发现,梯度条件能较理想的将物种五种物质分开,而在等度条件下无法实现,特别是蒽醌1,5-二磺酸钠和氨基萘酚-二磺酸钠,1-萘磺酸那和2-萘磺酸钠两组性能保留性能相似的物质较难分开。 谱分离图来看,三种芳环二磺酸盐和萘磺酸盐完全分开,分离效果较好。蒽醌1,5-二磺酸钠和蒽醌1,8-二磺酸钾因空间结构差异,所表现出来的保留性有明显的区别,因此可以较好的分离。1-萘磺酸钠和2-萘磺酸钠结构性质非常相似,化学性质也相近,较难分离,目前还未建立将两者完全分离的方法。图中所示分离度不如其他物质理想。
急需甲磺酸丁酯,哪位大虾可以指点从哪里可以购买到,急急!
求助!!老师让我找紫外中,萘环、磺酸基的特征峰分别是多少?找不到啊!求论坛大侠帮忙!!!跪求啊!!!
那位大侠知道[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析工业用环氧丙烷中杂质的方法?急。。
请教:丁二磺酸钠的定性定量方法
试问:硫酸铈能否氧化对叔丁基苯酚?可以直接电位滴定测对叔丁基苯酚的纯度吗?实验室电位滴定仪:梅特勒T50。若手动滴定,指示剂采用二苯胺磺酸钠还是邻二氮杂菲-亚铁?望指点
谁有氮-杂环丁烷纯度的检测方法采用液相检测,分享一下啊,急用,谢谢!!!
如题~最近使用mCPBA环氧化烯烃时,用二氯甲烷溶解,冰浴反应5min,室温放置10min后,加10%亚硫酸钠溶液洗涤离心,除去过量间氯过氧苯甲酸后,再用10%碳酸钠溶液洗涤,离心,再水洗,氮吹后上机分析,结果不太如意,进GC-FID分析后出现了更多杂质,mCPBA纯度70%,不知道问题出在哪,有没有做过此反应的大神,求指导!!!
二氧化硫甲醛吸收液中, 氨基磺酸和氨磺酸 一样吗? 氨基磺酸能代替氨磺酸吗?
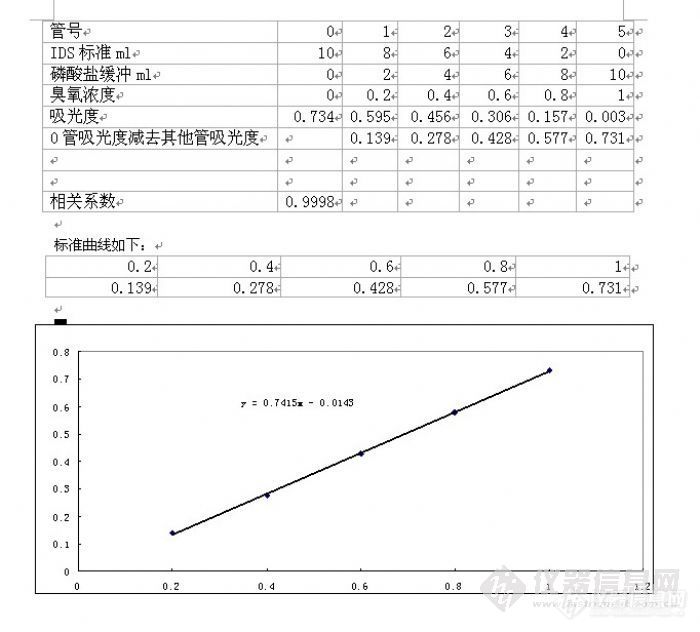
我用靛蓝二磺酸钠法测定空气中的臭氧,依据是GB/T 15437-1995修订后的国标方法,用0.25克靛蓝二磺酸钠配置储备液,进行标定后绘制的标准曲线的斜率总是0.74左右,达不到国标的要求,请问大家做的0.25克靛蓝二磺酸钠的浓度一般是多少,标准曲线斜率一般是多少?我做的总是达不到要求,是什么原因啊?请大家指教,不胜感激!![img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911212131_185820_1625938_3.jpg[/img]
按:烷基苯磺酸测定分子量,国标方法是用气相方法来做的。但是由于条件的限制,我们实验室没有办法测试。所以,退而求其次,使用滴定方法。发现使用滴定方法,能够为我们研发人员提供相对准确的帮助。LASH 相对分子量的测试 目的通过用标准氢氧化钠溶液滴定烷基苯磺酸的间接方法计算其分子量。原理R-SO3H + NaOH → R-SO3Na + H2O试剂原料0.5mol/L氢氧化钠标准溶液。锥形瓶中性乙醇方法称取5.0g样品到锥形瓶中,加入40ml中性乙醇,加热微沸。稍冷,加入4滴酚酞指示剂,用0.5mol/L NaOH 滴定液滴定至显出微红色,即为终点。计算公式 http://ng1.17img.cn/bbsfiles/images/2011/09/201109061142_314530_1626663_3.gif 其中:M——烷基苯磺酸的分子量(g/mol)m—样品称样量(g)c——NaOH浓度(mol/L)V——NaOH滴定体积(ml)α——样品的含量浓度(%)案例 序号123称样量5.12545.29135.3273滴定体积32.0033.0933.40分子量320.3319.7319.0备注样品含量为96%
现在做环境空气的氮氧化物实验,根据HJ479-2009中规定,对氨基苯磺酸溶解于200mL40到50度的热水中,请问大家做这一步时,药品都能溶解吗?我通常把水烧到50度,也不能充分溶解所有的对氨基苯磺酸,而且发现如果在溶解时,断断续续的加热,配出的显色液会变成淡红色。求大神分析下该怎么操作?还有就是,我配置的显色液并没有规范中说的在25摄氏度,暗处保存可稳定三个月,感觉过上1个星期,空白值就会越来越高。
各位高手,我想请教一下,测定食品中甜蜜素(环已基氨基磺酸钠)的毛细管柱;最好能 请提供样品(饮料、果汁、糕点)的前处理和色谱柱使用条件。谢谢了啊,在线等答复~~

氨基磺酸和氨磺酸虽然分子量相同,但结构不用,性质不同,我认为是两种不同的物质,但是市面上购买的氨磺酸全部发货氨基磺酸。我们以前用的是[img=,690,322]https://ng1.17img.cn/bbsfiles/images/2022/02/202202111509130721_3948_3036083_3.jpg!w690x322.jpg[/img]现在都是发的阿拉丁的氨基磺酸,再按照482-2009步骤配置氨磺酸钠结果会偏大百度上查了一下,也询问了一部分同行,有的说是同一种物质,有的说不是
有谁知道哪里卖环氧丁酸对照呢?
二氮杂菲测水中阴离子表面活性剂---定容某市环境监测中心 董捷 姜程程(图片无法显示,文章未完,请下载附件给予批评指正,谢谢)1. 范围本标准GB/T 5750.4-2006规定了用二氮杂菲萃取分光光度法测定生活饮用水及其水源水中的阴离子合成洗涤剂。本法适用于生活饮用水及其水源水中阴离子合成洗涤剂的测定。本法最低检测质量为2.5μg。若取水样100mL水样测定,则最低检测质量浓度为0.025mg/L(以十二烷基苯磺酸钠计)。生活饮用水及其水源水中常见的共存物质(mg/L)对本标准无干扰:Ca2+、NO3-(400)、SO42-(100)、Mg2+(70)、NO2-(17)、PO43-(10)、F-(7)、SCN-(5)、Mn2+、Cl2(1)、Cu2+(0.1)。阴离子表面活性剂质量浓度为0.1 mg/L时,会产生误差为-28.4%的严重干扰。2. 原理水中阴离子合成洗涤剂与Ferroin(Fe2+与二氮杂菲形成的配合物)形成离子缔合物,可被三氯甲烷萃取,于510nm波长下测定吸光度。3. 仪器3.1 分液漏斗,250mL。3.2 302B自动液液萃取仪。3.3 VIS-723G分光光度计。4. 试剂4.1 三氯甲烷4.2 二氮杂菲溶液(2g/L):称取0.2g二氮杂菲(C12H8N2•H2O,又名邻菲罗啉),溶于纯水中,加2滴盐酸(ρ20=1.19 g/mL),并用纯水稀释至100mL。4.3 乙酸铵缓冲溶液:称取250g乙酸铵(NH4C2H3O2),溶于150 mL纯水中,加入700 mL冰乙酸,混匀。4.4 盐酸羟胺-亚铁溶液:称取10g盐酸羟胺,加0.211g硫酸亚铁铵溶于纯水中,并稀释至100mL。4.5 十二烷基苯磺酸钠标准储备溶液:称取0.500g十二烷基苯磺酸钠(C12H25-C6H4SO3Na,简称DBS),溶于纯水中,定容至500 mL。4.6 十二烷基苯磺酸钠标准使用溶液:取十二烷基苯磺酸钠标准储备溶液(4.5)10.00mL于1000mL容量瓶中,用纯水定容。5. 分析步骤5.1 分别取0mL,0.5mL,1.00mL,2.00mL,3.00mL,5.00mLDBS标准使用溶液(4.6)于100mL容量瓶中,加纯水至100mL,混匀。5.2 按如下步骤进行实验操作。5.2.1 加入标准溶液于六个分液漏斗中分别加入配置好的100 mL DBS标准使用液,如图1。图1. 100 mLDBS标准使用液5.2.2 加入2 mL二氮杂菲溶液于六个分液漏斗中加入2 mL二氮杂菲溶液(4.2),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次将标准使用液与二氮杂菲混匀,如下图2。 图2. 二氮杂菲与标准液混匀5.2.3加入10 mL缓冲液于上述分液漏斗中再加入10 mL缓冲液(4.3),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次使其混匀,如下图3。 图3. 缓冲液混匀图5.2.4 加入1.0mL盐酸羟胺-亚铁溶液于六个分液漏斗中一次加入1.0mL盐酸羟胺-亚铁溶液(4.4),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次使其混匀,如下图4。 图4.盐酸羟胺-亚铁混匀图5.2.5 加入10mL三氯甲烷 于上述分液漏斗中加入10mL三氯甲烷,萃取强度设为50Hz,运行时间60s,停止时间30s,萃取两次使其混匀,如下图5。 图5. 三氯甲烷萃取图5.2.6 静置分层静置数分钟,使得三氯甲烷与水相分层,如下图6。 图6. 静置分层5.2.7 放液,定容 于分液漏斗颈部塞入一小团脱脂棉,慢慢旋开活塞,使得三氯甲烷成滴滴入干燥的10mL比色管中,并将少量三氯甲烷加入分液漏斗中清洗脱脂棉,并合此三氯甲烷并于比色管中,最后用三氯甲烷定容至10mL,混匀,以供测定。如图7所示。图7. 放液图5.3 于510nm波长,用3cm比色皿,以三氯甲烷为参比,测定吸光度。5.4 绘制工作曲线。未完,详细资料请下载附件,谢谢!
先说结论:衍生化后的环己烷氨基磺酸钠标准溶液在25摄氏度下5个小时就会有变化,主色谱峰[font=&]环己醇的面积会变小,衍生物色谱峰环己醇亚硝酸酯的面积会变大(应该是两者面积和不会变化,理论上还是可以继续用来做标准曲线的)。而且低浓度的环己烷氨基磺酸钠标准溶液衍生化后的色谱峰环己醇的色谱峰会出现杂峰。 我猜测高浓度的环己烷氨基磺酸钠标准溶液衍生化后的色谱峰环己醇的色谱峰不会出现杂峰是因为主峰比较大,把杂峰包裹进去了。 [/font] 上午对环己烷氨基磺酸钠标准溶液进行衍生化处理,浓度分别是10μg/ml、50μg/ml、500μg/ml,处理完毕后立刻上机。一共是12个进样瓶,第一次上机最后一个样品跑完后,大概时间是中午12:30,然后等待下午2:30的时候,又重新上机。 每个进样瓶的信号采集时间大概是20分钟,这样第一个样品和最后一个样品间隔时间是20*11=220分钟,加上中午的2个小时,这样大概是5个小时。也就是说第一批和第二批进样中每个相同序号的进样瓶采集时间间隔是5小时 这次使用的环己烷氨基磺酸钠标准溶液有两种,分别是10个月前配置的标准溶液和新配置的标准溶液。 分别编号是10-1、50-1、500-1、10-2、50-2、500-2 下面是各个对比的色谱图,分别是上午的色谱图和下午的色谱图进行对比 10-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇旁边出现了杂峰 [img=,690,373]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124064647_4366_5979722_3.png!w690x373.jpg[/img] 50-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,457]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124061287_876_5979722_3.png!w690x457.jpg[/img] 500-1的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,500μg/ml的这个浓度上午和下午的色谱图区别不大 [img=,690,446]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181124064695_5130_5979722_3.png!w690x446.jpg[/img] 10-2的对比,上午和下午的色谱图进行对比,环己烷氨基磺酸钠标准溶液的主色谱峰旁边出现了杂峰 [img=,690,413]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150579387_7325_5979722_3.png!w690x413.jpg[/img] 50-2的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,481]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150587943_3504_5979722_3.png!w690x481.jpg[/img] 500-2的对比,上午的色谱图几乎正常,下午再跑一次的色谱图,主色谱峰环己醇面积明显变小了,衍生物色谱峰环己醇亚硝酸酯的面积明显变大了。 [img=,690,509]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181150579543_2309_5979722_3.png!w690x509.jpg[/img]
请问2mmol庚烷磺酸钠作为定容液能进质谱吗?只是作为定溶液上机用,进样量为20μL,做氨基糖苷类,不想用七氟丁酸。但是又担心庚烷磺酸钠作为离子对试剂会对MS有影响。
最近有个朋友要测试全氟丁基磺酸钾,让我帮忙问问[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]能不能测试,分子量有点大,保留应该比较强,大家有没有人做过?
现在合成一个化合物 萘环左边环有一个磺酸基,右边环1位是酚羟基 2位是氨基,对这个化合物液相分析,流动相PH4,出现两个峰,固体样品溶解后进样 前面峰是主要的峰,但是样品放了一会后再进样 就变成后面是主要的峰了,请问这两个峰都是我的产物么,这个化合物本身会有离子分子间转换么 ,查到有苯类带磺酸基和氨基说会形成内盐。
正在研究丁二烯的过氧化反应,产物为3,4环氧-1-丁烯。由于色谱仪中只能使用填充柱而且最高温度不能超过60度。之前使用过17%的Carbowax作为固定液,不能分离丁二烯和3,4环氧-1-丁烯。不知大家有没有这方面的经验,希望告知,谢谢了!
谁有丁二酸酐,三乙胺,对甲基苯磺酸的检测方法

遗传毒性杂质,现在是药学研究的焦点之一。甲磺酸、苯甲磺酸等磺酸盐类物质与微量的低级醇在合成反应中生成烷基磺酸酯类,这些物质可与DNA发生烷基化反应,从而可能成为引发癌症的诱因。欧洲医药评价署、美国食品和药品管理局及国际药品注册协调会议等先后对基因毒性杂质做出限度规定。具体到我们的甲磺酸加贝酯产品,需要对其中的甲磺酸乙酯的限度进行控制。溶液制备:对照品溶液 取甲磺酸乙酯适量,精密称定,用乙腈制成每毫升含1.5μg/ml的溶液,精密移取2ml加入顶空瓶中,加入3ml水,6g碘化钠后,扎盖密封。供试品溶液 取甲磺酸加贝酯适量,精密称定,按供试品100mg与乙腈1ml的比例配制供试品溶液,溶液经超声、波膜过滤处理后,精密移取2ml加入顶空瓶中,加入3ml水,3g碘化钠后,扎盖密封。色谱条件:Agilent 7890色谱仪,顶空进样器,FID检测器;色谱柱,月旭WEL-PEG20M,30m*0.32mm*0.25μm(Cat. NO:01918-32001;Ser. NO:GC20131102);进样口温度为110℃,检测器温度为260℃,氢气流速为30ml/min,空气流速为350ml/min,进样量为1mL,分流比为0.1:1。升温程序,起始温度为40℃,维持10min,然后以20℃/min的升温速率,升温至160℃,维持1min。顶空瓶平衡温度为80℃,平衡时间为30min。结果:对照液色谱图: http://ng1.17img.cn/bbsfiles/images/2014/07/201407021418_503868_1609327_3.jpg其中,时间为1.982min的保留峰为甲磺酸乙酯的衍生物。供试液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/07/201407021538_503896_1609327_3.jpg由色谱图中可以看出,样品中未检出甲磺酸乙酯。讨论:出峰时间非常的快,但是理论塔板数、分离度、对称因子等却非常给力!既得到了良好的分离效果,又尽可能的节约了分析时间。







 我要推广仪器
我要推广仪器
 下载APP
下载APP