求购R、S-四氢呋喃甲酸[em01] [em01] [em01]
打算用《GB T 20388-2016 纺织品 邻苯二甲酸酯的测定四氢呋喃法》,做DBP 的单标 空白样品加标回收率实验,请问1:应该加哪几个浓度的DBP 单标?(检出限是0.4mg/L)2:加多少体积的DBP 单标3:是否需要加内标谢谢

请教各位老师:采用四氢呋喃超声萃取邻苯二甲酸酯进样测定数据与实际差距较大,是哪块步骤出了问题?0.3g样品+10ml四氢呋喃超声30-60min,加入20ml正己烷静置,聚四氟乙烯膜过滤。样品为邻苯浓度1000ppm的PVC,实测只有5ppm,定量方法为外标法,同一方法参数对5ppm、10ppm等浓度标液进行检测无异常。[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2023/07/202307161725554321_1260_5881265_3.jpg!w690x920.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/07/202307161725556394_1136_5881265_3.jpg!w690x517.jpg[/img]
[font=&]打算用《GB T 20388-2016 纺织品 邻苯二甲酸酯的测定四氢呋喃法》,做DBP 的单标 空白样品加标回收率实验,请问1:应该加哪几个浓度的DBP 单标?(检出限是0.4mg/L)2:加多少体积的DBP 单标3:是否需要加内标谢谢[/font]

想开展 邻苯二甲酸酯的测定 四氢呋喃法这个方法分析,现在实验室只有GCMS ,还需添加什么设备吗?[img=,690,126]http://ng1.17img.cn/bbsfiles/images/2018/05/201805091005101776_5535_2154459_3.png!w690x126.jpg[/img]
呋喃的方法扫了好几次改了各种参数,好不容易是有一点响应了,0.5的点只有700多,进样量10微升,但是进样5微升0.5的话基本是不出的,改了很多参数(加热器和干燥气)流动相是乙腈和0.1%甲酸加1mol的甲酸胺,希望前辈能分享一下呋喃响应低的解决办法。
检测有机磷,氨基甲酸酯类、磺胺类、呋喃类,氯霉素类、红霉素、青霉素等,用什么色谱柱?
GBT 20388-2016 纺织品 邻苯二甲酸酯的测定 四氢呋喃法,大家有做的吗,是不是可以和偶氮共用一个仪器呢?
求助无水硫酸镁,氢化钠,氯甲酸乙酯,四氢呋喃,乙腈(hplc),N-甲基吗啉,二异丙基乙胺,对甲苯磺酸的质量标准。大家有哪个给我哪个就可以。这些资料库里都没有。在外面也实在找不到了。大家帮忙。
请教各位行家,最近开始使用Aglient 5973N型GC/MS,在检测氨基甲酸酯类农药:速灭威、抗蚜威、呋喃丹时配置的混标浓度均为4ug/ml,仪器设置的条件inlets:250度 split 20:1 OVEN:130度保留时间2min每分钟升15度升到220度。运行15分钟。柱:HP-5MS 30.0m*0.25mm*0.25um 可是根本就检测不到,这是什么原因啊?,还有,好象农药库里面根本就没有速灭威(tsumacide)这种农药是吗?那要怎样才能够定量呢?谢谢
小弟 最近在用6460C测定硝基呋喃 加入20毫升 0.2mol/L的盐酸 加入内标1ug/mL 混表1ug/mL 各0.5毫升 于50毫升离心管(外壁用锡纸包裹)衍生试剂 0.5毫升(二甲亚砜溶解 0.075g到10毫升) 混匀后 37℃水浴过夜 第二天取出加入 磷酸二氢钾 5毫升 使用氢氧化钠溶液调节溶液PH在 7.4左右 之后使用15毫升乙酸乙酯分三次提取 收集乙酸乙酯层到氮吹管中 40℃氮吹干 后使用甲醇定容 1mL 后上机测试 流动相是 0.1甲酸水 和乙腈 经过多次测定 发现没有离子对出现 没有出峰 想向大神们求助 这个项目该如何做
我最近用国标21311法做动物食品中硝基呋喃检测,按方法衍生后上机,只有AOZ和AOZ内标出峰正常。AHD,SEM不出峰。方法优化时,AHD和SEM的响应很差,我的流动相用的0.1%甲酸水和乙腈。有没有做过的老师指点一下是哪里的问题,谢谢。
国标测硝基呋喃,加磷酸钾的意义是什么?调PH? 什么原理求解答? 谢谢
最近用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做硝基呋喃类不出峰液相洗脱条件是参照农业部783-1-2006标准的A是乙酸铵,B是甲醇我用的是A是0.1%甲酸水,B是已腈完全不出峰衍生是用前处理盒衍生的大神帮我看看是流动相问题,还是梯度设置问题[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808171134395095_4831_3099829_3.jpeg[/img]
我的样品分子量在2000以下,流动相用进口的四氢呋喃。每次新开封一瓶四氢呋喃后,我会倒一部分四氢呋喃到一个棕色瓶中,用于平时试样的配制。但我发现,几天后棕色瓶中的四氢呋喃就氧化了,氧化物的分子量也在2000以下,出峰正好对我的试样形成干扰了。请教大家有什么好的防止四氢呋喃氧化的方法?还是我每天制样时,必须从流动相那个瓶中抽取四氢呋喃,才能减少这种干扰?
肌肉中硝基呋喃类代谢物的前处理检测1、洗涤;称样2g,加入50%甲醇水10ml.震荡后4000r/min,离心5min.洗涤两次后,弃上清液。2、衍生 ;混合内标标液20ng/ml,加入样品后,加入0.1ml0.1mc/lml2-硝基苯甲醛溶液,加入0.2mc/lml盐酸溶液10ml.37℃避光水浴16h,冷却至室温(避光)。3、提取 用 1.0mcl/ml磷酸三钾调PH值至7.0,加入10ml乙酸乙酯,混匀震荡5min后,6000r/min离心10min,取上清液50℃水浴氮气吹干后,用1.0mc/lml0.1%甲酸溶液1.0ml溶解残留物,过滤,上机测定。
我做硝基呋喃检测时做添加,呋喃西林和呋喃妥因的响应值比较低,请教各位高手是哪有问题呀?多谢!!
新手小白,刚接触[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],想请教走呋喃西林代谢物时,用的乙腈,0.1%甲酸水作为流动相,在4分钟左右出峰,响应值不高,离子对209-192和209-166,192碎片离子几乎是没有丰度的,而且调节它的碰撞能量也没有反应,请问这是什么原因呢?
大家好: 我在做硝基呋喃代谢物时遇到以下困难请大家指点: 当流动相为甲醇+0.5mmol乙酸铵时呋喃妥因代谢物后面有个包(应该是杂质干扰),当流动相改为甲醇+5mmol乙酸铵时呋喃西林代谢物的噪音有很高,请问大家有没有遇到这种情况,请给予指点,谢谢!
硝基呋喃类呋喃西林响应值不好,呋喃妥因,呋喃它酮线性不好,然后唑酮没有问题,混标,不知道哪个地方出问题了,请教一下各位老师。
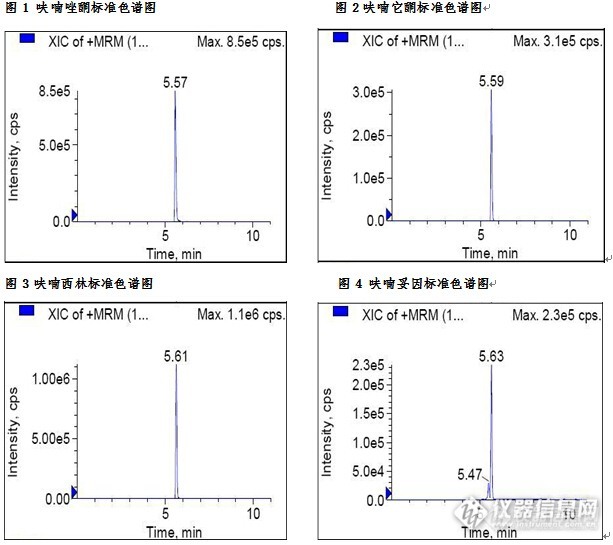
动物源性食品中硝基呋喃类药物检测的固相萃取方法一、实验目的本实验利用固相萃取法作为样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂用量。二、实验目标物呋喃唑酮(CAS:67-45-8),呋喃它酮(CAS:139-91-3),呋喃西林(CAS:59-87-0),呋喃妥因(CAS:67-20-9)。三、应用范围本方法适用于动物源性食品中硝基呋喃类药物的LC-MS/MS检测及确证。四、参考文献 推荐性国家标准《GB/T 21311-2007 动物源性食品中硝基呋喃类药物代谢物残留量检测方法 高效液相色谱-串联质谱法》。五、实验材料 C8/SAX固相萃取柱200mg/6mL。六、实验方法1、样品前处理 将样品组织搅碎、均质。精确称取约1g(精确到0.01g)样品于15mL带螺盖的离心管中,加入1mL水和8mL甲醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙酸乙酯,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液。2、水解和衍生化向质控和样品同待测样品中加入5mL 0.2mol/L的盐酸水溶液、100μL衍生化试剂、以及100μL内标工作溶液(20μg/L)和的混合标准溶液(10μg/L)。盖好盖子,充分振荡混匀,然后放入空气浴摇床,在37℃,200r/min条件下衍生化16h(过夜)。3、样品提取 加入0.3M的磷酸钠水溶液500μL。用试管滴加10mol/L的氢氧化钠溶液,涡旋混合均匀,用精密pH试纸调节pH值至7.0—7.2。9500r/min 离心10min,取上清液。4、SPE柱净化(1)活化:依次以5mL甲醇和5mL纯水预处理。(2)洗脱:上清液全部过柱,流速控制在约每秒1滴。依次以5mL水和5mL 50%甲醇/水溶液淋洗柱子。淋洗液完全通过小柱后,至少抽真空5min。以4mL 4%的甲醇氨洗脱,洗脱液用15mL试管收集。(3)浓缩定容:40℃氮气吹干。残渣以0.05%甲酸/甲醇溶液(9:1,v/v)溶解。溶解液以0.22μm的水相滤膜过滤,滤液可直接用于LC-MS/MS分析。5、LC-MS/MS条件 液相色谱-质谱/质谱仪 色谱柱:C18柱:150mm×2.1mm,2.0μm,或相当者 流动相:甲醇-5mM乙酸铵七、实验结果1、添加回收结果 向样品中加入不同水平的四环素类药物,回收率结果如下:(见表1)表1 动物组织中四环素类药物添加回收结果 样品名称 化合物名称 添加水平(ng/mL) 回收率(%) 猪肉 呋喃唑酮 50 80.75 100 82.58 呋喃它酮 50 89.74 100 90.88 呋喃西林 50 92.74 100 91.28 呋喃妥因 50 95.63 100 96.94 2、 空白样品添加农药残留物色谱图 http://ng1.17img.cn/bbsfiles/images/2015/08/201508141653_560805_3310_3.jpg
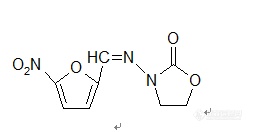
【中文名称】呋喃唑酮;痢特灵;3-(5-硝基呋喃甲叉)胺基-2-噁唑烷酮【英文名称】furazolidone;foroxone【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203012030_351889_1855403_3.jpg【性状】 黄色结晶性粉末,无臭味苦。【溶解情况】 微溶于水和乙醇。【用途】 抗菌谱类似呋喃因。对大肠杆菌、痢疾杆菌等最敏感。但口服时肠道不易吸收。用于菌痢和肠炎。也可用于泌尿道感染。 可作饲料添加剂,较早应用于防治球虫病,主要对球虫生长过程中的第二代繁殖体有作用,可预防和控制鸡脆弱艾美耳球从、毒害艾美耳球虫、堆形艾美耳球虫,除对球虫原虫作用外,对细菌性痢疾也有效。。【制备或来源】 (1)可由β-羟基乙基肼与碳酸二乙酯经环合、缩合制得。 (2)由乙醇胺经缩合,亚硝化,环合,还原,再缩合制得。【生产单位】略
样品制备 制备方法:标准品衍生方法:取硝基呋喃代谢物混标200 μL,加入2 mL 0.2 mol/L 盐酸溶液和20μL 衍生剂,混合后置于37℃条件下保持4 h。注:0.2 mol/L 盐酸溶液:量取17 mL浓盐酸,用水定容至1000 mL。衍生剂:含2-硝基苯甲醛0.05 mol/L。称取0.075 g 2-硝基苯甲醛溶于10 mL二甲基亚砜,现用现配。分析条件 色谱柱:EndeavorsilTM C18 ,100×2.1 mm,1.8 μm(Cat#:87003) 流动相:A: 0.4% 甲酸水B:乙腈流速:0.2 mL/min柱温:30 ℃检测器:UV 254 nm进样量:2.0 μL梯度洗脱表时间(min)A:0.4%甲酸水B:乙腈07030370303.0120808.0020808.01703015.007030http://www.dikma.com.cn/Public/Uploads/images/2(112).PNGhttp://www.dikma.com.cn/Public/Uploads/images/3(89).PNG
问题: 请教下各位老师,那个硝基呋喃代谢物一直没做好,四种物质都出来了,有两种响应值特别的高,有两个基本上没什么响应值,这个是什么原因?标准品,衍生剂,条件都控制好了。这个问题困扰我很久了,希望大家帮我解决下回复: AHD和SEM本身响应值就低,买高端一点的质谱,灵敏度更高的
最近在做食品中硝基呋喃代谢物时,发现四种硝基呋喃代谢物中,三种标线都没有问题,只有AHD的标线超差,差的原因为标线最后两个点的内标峰面积突然增大很多,导致内外标峰面积比值的增加与浓度增加不成比例,这是为什么呢?外标是成线性的。
问题: 你们谁硝基呋喃有检出过的?回复1: 我用的是[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质测的,不是[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]质,这个方法不行啊[难过][难过]回复2: 我们做硝基呋喃。。ahd的子离子有事信号几百,有时几千。。一个样都是这样。。不知道怎么回事
做卡托普利时,按照标准配制磷酸盐和甲醇的四氢呋喃的混合溶液作流动相,但头天晚上和第二天早上主峰的保留时间相差较大,请问各位在工作中有没有遇到过加四氢呋喃的流动相?会影响保留时间吗?(之后没按标准来,直接四氢呋喃占个通道运行,保留时间就未再漂移了。)
各位大侠,我在做水质呋喃丹分析时,按照国标GB/T5750-2006农药指标方法15.1液相色谱法做水中呋喃丹,色谱柱为安捷伦ZORBAX C18 150mm*4.6mm*5um,条件和国标一致,为什么不出峰,标准系列最高点100ug/l都不出峰。请教各位用的是什么柱子,具体条件是什么?我从文献上看的是C8的柱子,不知各位大侠用的是什么条件?谢谢
10号走呋喃代谢物样品的时候,发现了一个很奇怪的现象,标准品保留时间正常,走空白和样品时AMOZ、SEM、AHD、AOZ四项内标保留时间全部偏离0.2min左右,最后一针标准品也没能幸免。可以肯定不是流动相的问题,柱子平衡时间也足够长。为了确认,又重新进了一遍,仍然重复上述情况。最后,把空白舍去,按照标准品、样品、标准品顺序运行,结果正常,空白的影响力这么大?一针空白可以导致后面序列全部异常(总共6针)?我真是第一次碰到这种奇怪的现象,问题的原因还没有找到,等看一下今天的样品结果再做判断吧。仔细看了一下,发现空白没有内标,测了一下空白的PH,大约在5-7,标准和样品都在3左右,难道真的是弄错了空白?查看了当天的样品,同时检测的还有克伦特罗、孔雀石绿、氯霉素和盐霉素,前三个都有内标监控,所以肯定没错,最有可能的是和盐霉素空白混了,想重新检测比较一下,结果进样小瓶已经处理掉了,哎,只能等有时间再验证了。17和18号进行了相关验证,发现使用盐霉素空白并不能导致呋喃代谢物保留时间漂移,看来当时的呋喃空白应该是预处理操作的时候出现了错误,因无法重现,可能要成为不解之谜了。。。。。。以下是当天保留时间偏离的三个样品和标准的TIC图谱,其中第一、三、五、七通道分别是AMOZ、SEM、AHD、AOZ的内标,每一通道前三个是样品,最后一个是标准品。大家看一下有没有遇到这样偏离的情况,讨论一下。http://ng1.17img.cn/bbsfiles/images/2010/11/201011202027_260858_1855403_3.gif
请问要测定呋喃树脂的粘度选用什么型号的粘度计比较适宜要测粘度单位Pas,谢谢各位赐教!







 我要推广仪器
我要推广仪器
 下载APP
下载APP