最近做硫化氢实验遇到一个问题,配置混合显色剂的三氯化铁凝固了,我放在水浴锅上加热(没碰到水),三氯化铁就变成溶液了,然后我称了50g配成50ml的三氯化铁溶液,与NN二甲基对苯二胺盐酸盐使用液配成混合显色剂,请问这么做对实验有没有影响
请教水质 色度 六氯铂酸钾和六水氯化钴用哪家的试剂呢?用分析纯还是优级纯呢?配标准色度用的
ph测试中的,三级水的结果跟氯化钾的结果经常有很大差异,甚至1点几,感觉不正常,有可能是哪里出了问题呢?
三氯化六氨合钴哪个厂家的好?有人推荐进口的,10g要七八百元,有国产品牌的吗?推荐一个。

购买的三氯化六氨合钴物质,进口的,10g,用的时候都不舍得。[img=,690,1226]https://ng1.17img.cn/bbsfiles/images/2019/06/201906111813251164_8292_2826867_3.jpg!w690x1226.jpg[/img]
三氯化铁和硫酸铜在液相条件下能出峰吗? 谢谢色谱柱 C18流动相 乙腈: 缓冲液 (10%四丁基氢氧化铵溶液6ml,加水至200ml,磷酸调节PH为6.5):水(20:20:60)检测波长254NM,流速1请问在这种体系 下 硫酸铜和三氯化铁 会出峰吗:为什麽?
用三级水和氯化钾测试ph值结果相差多少啊?
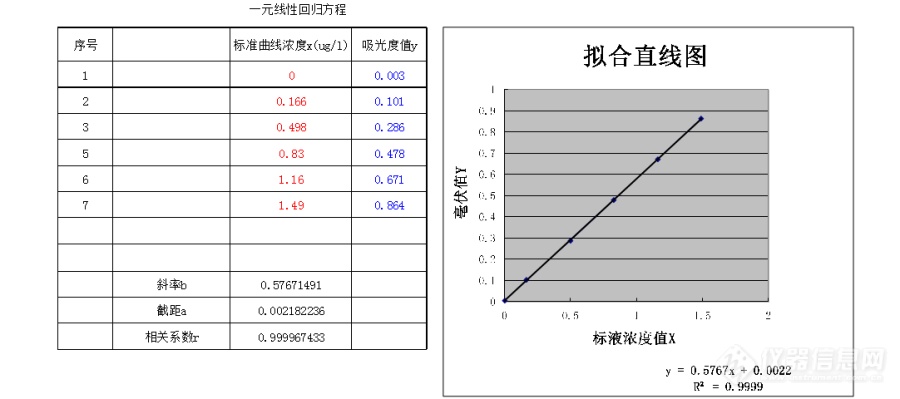
用HJ889-2017三氯化六氨合钴测土壤中阳离子,标准曲线如下,样品土分别是1.5、0.8、3.6,土壤PH值是7.55,7.91。[img=,690,305]https://ng1.17img.cn/bbsfiles/images/2019/06/201906041109194001_9498_1645480_3.png!w690x305.jpg[/img]
RT,做三氯化磷,三氯化氧磷,五氯化磷,三氯硫磷,磷酸三乙酯的朋友有没?本人QQ444980036大家一起聊聊啊,最好是质检方面的,嘿嘿!!
我们最近扩项要做植物油碘价.可是我对标准里一个试剂配制产生问题: 根据在网上下载的标准GB/T5532-1995--"5.5试剂(含一氯化碘的乙酸溶液):称9g三氯化碘溶解在700mL冰乙酸和300mL环己烷的混合液中。 取5mL上述试剂加5mL碘化钾溶液和30mL水,用几滴淀粉溶液做指示剂。用0.1mol/L硫代硫酸钠标准溶液滴定析出的碘,滴定体积V1。 加10g纯碘于试剂中,,使其完全溶解。如上法滴定,得V2。V2/V1应大于1.5,否则可稍加点纯碘直至V2/V1略超过1.5。 将溶液静置后将上层清液倒入具塞棕色试剂瓶中,避光保存,此溶液在室温下可保存几个月。" 其中试剂5.5就是后面分析步骤中提到的wijs试剂. 但是,我在网上直接搜"wijs试剂"得到的配制方法是-- " 取6~7mL纯一氯化碘溶解在1L冰醋酸中,暗处保存" 那我要做碘价分析,应该按哪种方法来配呢?还是标准中有错误,"9g三氯化碘"应该为"9g一氯化碘"[em06]
本人新手,刚开始做三氯甲烷四氯化碳,标液是新拆的,仪器各项条件不动,用的之前同事调好的,稀释标液用的水是蒸馏水煮沸20分钟,水浴平衡1小时,配的混标,做出来的峰面积与之前做的差五六百倍,而且三氯甲烷线性良好,四氯化碳不成线性,是什么原因啊注:手动顶空进样,进样60uL
我们实验室用国产纯水机处理过的水中三氯甲烷、四氯化碳含量都挺高,煮沸十分钟冷却后再测,空白还是含大家都是用的什么水?有没有直接卖的比较好用的?上超市买过哇哈哈什么的,效果也不好

按照HJ 620-2011 方法,顶空-ECD做水中的14种挥发性卤代烃类。早先就听过版友们说过,纯水机制出的水做这个项目空白是不行的(好像是超市里几乎所有的瓶装水都是不行的——因为这些水都是塑料瓶)。于是就提前准备,买了一瓶屈臣氏的蒸馏水。然后按照标准做实验。结果。。。发现三氯甲烷和四氯化碳还是杯具的检出了。http://ng1.17img.cn/bbsfiles/images/2014/04/201404081704_495657_2206495_3.jpg哎,这ECD检测器也太灵敏了,于是拿这瓶屈臣氏蒸馏水再去重新蒸馏一下吧。。。全玻璃蒸馏器。http://ng1.17img.cn/bbsfiles/images/2014/04/201404081701_495655_2206495_3.jpg取500毫升水蒸馏,弃去前150毫升和后150毫升水,然后拿着蒸馏出来的水再上顶空做,结果。。。三氯甲烷仍然有检出,四氯化碳倒是少了。哎。。。这ECD检测器也太灵敏了。现在正在试着把蒸馏后的水先加入几十克经过350度灼烧过的优级纯氯化钠,然后再放到水浴锅里水浴,顺便再用氮吹仪吹着氮气在上边避免污染。http://ng1.17img.cn/bbsfiles/images/2014/04/201404081721_495659_2206495_3.jpg还想试试看看能不能解决这个空白吧。氮吹水浴了两个多小时,水也蒸发的不少了。再放到顶空瓶里一试,我勒个去。三氯甲烷还是很大再搞不定就真的没辙了。第二天再接着做。第二天,为了排除实验室空气的干扰,特地跑到平时做臭气浓度的房间里蒸馏了500毫升水,(水源是纯水机制的水,因为纯水机出了毛病,电阻率只有2)看看。因为楼下一位版友说可能三氯甲烷和四氯化碳会跟着水蒸馏出来,于是只蒸半瓶水,分别进样。结果发现不管是新蒸馏水还是蒸馏瓶里剩下的水,三氯甲烷都悲剧的检出了。但是都不高。中午跑到超市去买了一瓶法国依云的矿泉水(12块8,500毫升,真贵)又顺便买了一瓶最近广告上很火的恒大冰泉。再来一次试试吧。http://ng1.17img.cn/bbsfiles/images/2014/04/201404091415_495729_2206495_3.jpg
仪器:岛津2010PLUS,ECD检测器、Dani86.50顶空自动进样器色谱条件:色谱柱:RTX-1 30M*0.25mm*0.25um 柱温:60℃ 5min 进样口温度:200℃,检测器:250℃ 分流比:30 柱流速:1.5ml/min顶空条件:平衡温度:50℃,平衡时间:25min,六通阀温度:100℃,传输线温度:100℃今天做水中三氯甲烷、四氯化碳时,标准系列(ug/ml):三氯甲烷(0.02、0.05、0.1、0.15、0.2、0.5和四氯化碳(0.002、0.005、0.01、0.015、0.02),三氯甲烷和四氯化碳都在第四个浓度时色谱峰出现分叉。仪器是新的,色谱柱也是新的。
各位老师们,泰克玛顶空里残留三氯甲烷四氯化碳可以多少度加热去除?已经拆过了进样针和定量环甲醇超声半个小时烘干安装还是不好用,或者还有哪里隐藏三氯甲烷,四氯化碳,空白水是刚开封的娃哈哈,顶空瓶子都是新的
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]同时检测水的四氯化碳、三氯甲烷、四氯乙烯、三氯乙烯怎么配置混标?
[color=#444444]最近在做水中三氯甲烷四氯化碳的测定实验,参照标准GB/T 5750,仪器型号岛津GC-2010 Plus,配的是AOC-5000顶空自动进样器,其他条件如下:[/color][color=#444444]1.色谱柱:RTX-5 30*0.25*0.25的;[/color][color=#444444]2.顶空条件:40℃,振摇1小时,针45℃,进样体积300微升;[/color][color=#444444]3.仪器条件:进样口200℃,分流衬管,无石英棉;ECD检测器,250℃;柱流速2 ml/min,分流比20:1;柱温60℃,保留5min。[/color][color=#444444]4.色谱柱老化至300℃,检测器检查过,无污染,衬管是新的,垫片是新的,进样针是新的,试验中用的所有玻璃器皿,量器,包括顶空瓶,枪头,全都烘过。[/color][color=#444444]5.配制标线用水为屈臣氏蒸馏水,煮沸15分钟以上,煮水的屋子并非配制标准溶液的屋子。[/color][color=#444444]6.标准溶液配制时,在通风橱里。[/color][color=#444444] 目前出现了好多问题:[/color][color=#444444]1.走空针,不进样走一针,基线很平,噪音50多。[/color][color=#444444]2.走空气,标样配制室的空气,有干扰,三氯甲烷峰面积8000左右,四氯化碳6000左右。[/color][color=#444444]3.走屈臣氏水,即用来配制标准溶液的水,有干扰,三氯甲烷峰面积13000左右,四氯化碳6000左右。[/color][color=#444444]4.想配制标准曲线,扣除水的本底值,结果两种物质均仅在低浓度时成线性。即三氯在0.1-1.0μg/L,四氯化碳在0.01-0.1μg/L条件下成线性,相关系数可达3个9,。[/color][color=#444444] 那么问题来了:[/color][color=#444444]1. 各位大神们做的时候本底值也是这么高么?如何解决本底值高的问题?[/color][color=#444444]2.亲们做的检出限分别是多少?线性范围分别是多少?相关系数能达到多少?[/color][color=#444444]3.大神们一般都测定的什么样品?大概含量都是多少?[/color]
大家在依据GB/T5750.8-2006(顶空+ECD)检测三氯甲烷和四氯化碳时,有没有直接购买瓶装饮用水来代替实验室纯水?什么牌子的比较好?我用依云矿泉水试了一下,新开瓶的依云水还是能够检出一点三氯甲烷和四氯化碳。之后把依云水煮沸,趁沸腾时,直接取水样装入顶空瓶,上机检测,三氯甲烷和四氯化碳的浓度更大了,不知道是为什么,挺奇怪的。
请教各位大侠:我用安捷伦7820a色谱仪,做的三氯甲烷和四氯化碳曲线(0.2,1,2,4,6,10系列)老是刚过两个9,达不到三个9,不知原因是什么?蒸馏水也煮沸30分钟了,顶空自动进样时平衡时间是60度30分钟。。空白始终有点三氯甲烷和四氯化碳的峰,使用娃哈哈纯净水也是如此。不知再怎样处理?
用usp方法做六水合氯化镁定量分析 做出来总是101.9%,超出标准。如果是无水氯化镁就是99.9%. 就合格。太奇怪了。无水和六水合样品有何区别?六水的换了三个公司的试剂结果都一样
我是新手 所以 悬赏的少 希望你们见谅 但是我真的很着急 考上岗证 给我三个不好做的项目 硫化物 氯化物 总氮 我正式工作刚半年 实验经验少 看网上说硫化物和总氮都是属于不稳定的项目 很着急 标样是不会告诉大约值的 所以浓度大约多少也不知道 硫化物用的是亚甲蓝法 氯化物用的是硝酸银滴定法 总氮用的是碱性过硫酸钾消解法 其中有很多的问题我不懂 1.比如 硫化物标样和标液用什么水稀释比较好 2 标液一般都是取多少定容到多少 3 标样一般是取多少稀释到多少 再取多少? 4他们有说水得是碱性水 碱性水要怎么配 ?实验怎么避免硫化物挥发 很多细节我一时想不到 希望做过的哥哥姐姐叔叔阿姨们告诉我 总氮我们单位不会现给买进口的过硫酸钾怎么办 这个我知道对过硫酸钾要求很高的 空白不能大于0.03 氯化物 我都不知道要问什么好 你们把你们在实验中总结类似的经验教教我 好怕考不过啊
尾气吸收瓶,水吸收氯化氢和二氧化硫,其中主要是氯化氢,请问我这样计算有无问题,如果不对,该怎么计算呢?
[font=宋体]采用[/font]GB/T6730.65[font=宋体]三氯化钛还原[/font][font=宋体]重铬酸钾滴定法,分析铁矿石中全铁含量。[/font][font=宋体]测试样品使用的是中实国金质控样,指定值为[/font]66.84%[font=宋体],证书提示稳健标准差[/font]0.16%[font=宋体]。[/font][font=宋体]所有仪器均经过校准。药品使用均为西陇或者科隆的分析纯或基准级。用水采用[/font]GB/T6682[font=宋体]中规定的三级水,重铬酸钾[/font]2.4515[font=宋体]克直配法(标准中[/font]4.26[font=宋体]条),用[/font]1[font=宋体]级水配置。[/font][font=宋体]分析过程简述:差量法称取[/font]0.2000g[font=宋体]铁矿石,使用[/font]250ml[font=宋体]锥形瓶,微量水润湿矿样,加入[/font]15ml[font=宋体]氟化钠和[/font]20ml[font=宋体]浓盐酸,置于电热板上,控制温度在[/font]60-70[font=宋体]度。溶解[/font]10[font=宋体]分钟后,开始滴加氯化亚锡和盐酸(氯化亚锡还原三价铁离子,盐酸控制酸度),保持溶液黄色或淡黄色。随着矿石的溶解,溶液中三价铁离子会变多,因此需不时滴加氯化亚锡和盐酸,让新施放的三价铁离子被还原成二价,始终尽量保持溶液淡黄色。[/font][font=宋体]溶样完毕后(耗时[/font]1.5-2[font=宋体]小时),开始浓缩。浓缩温度在[/font]70[font=宋体]度左右,过程中液体减少,残余的三价铁离子变浓,因此溶液又开始变深黄。此时再分次滴入了少量氯化亚锡和盐酸,让溶液保持淡黄色,直到液体浓缩至约[/font]20ml[font=宋体]。[/font][font=宋体]冷却至室温后,加[/font]100ml[font=宋体]水,[/font]1ml[font=宋体]钨酸钠,晃动状态下滴加三氯化钛。此时三氯化钛只是解决掉没有被二氯化锡还原的残存三价铁离子。当还原完所有的三价铁离子,三氯化钛再过量一滴,即可和钨酸钠显色,成为钨兰。此时溶液中可被氧化的三价铁离子,视同被置零了,均转化为[/font]2[font=宋体]价。三氯化钛用量不大,几滴即出现钨兰。[/font][font=宋体]放置上述液体于空气中,待钨兰刚消退,立即加入硫磷混酸[/font]10ml[font=宋体](硫酸调节酸度,磷酸提供配位掉三价铁离子和降电位),加入二苯胺磺酸钠后,立即开始滴定,溶液从铬绿到转变为紫罗兰。读取滴定体积后,根据公式进行全铁含量计算。[/font][font=宋体]开展方法验证[/font]5[font=宋体]天,[/font]10[font=宋体]批以上,双样平行度很好,均在[/font]66.52-66.59%[font=宋体](低于指定值[/font]66.84%[font=宋体])之间,但是已经超了标准中规定的[/font]0.7R[font=宋体],即[/font]0.28%[font=宋体]。未分析出走了负误差的原因。[/font]1[font=宋体],请教偏倚都是低的原因所在?[/font]
三乙胺怎么除水呢,我要求不高,查资料说不能用硫酸镁,可以用氯化钙吗
我们实验室的纯水做空白,四氯化碳和三氯甲烷都很高,买了依云水、和葡萄牙的进口水都不行。也做了煮沸,活性炭吸附都不行。实验室的空气干扰可排除!我们进的第一针为甚目标化合物的出峰时间总是会变,之后又会和标准物质的出峰时间相同。
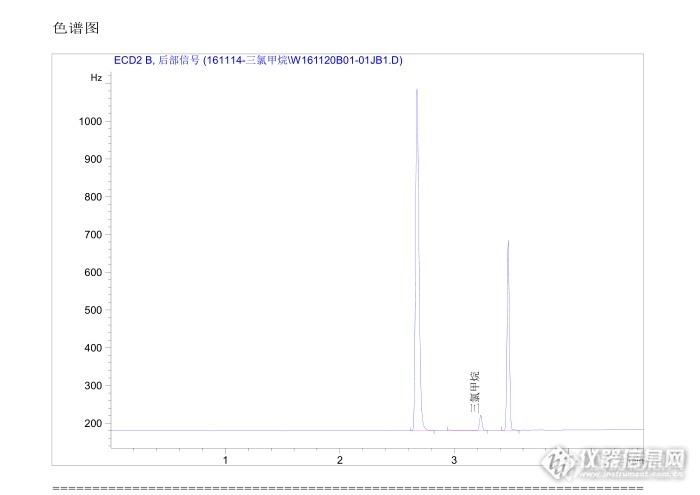
方法:GB/T 5750.8-2006 毛细管柱色谱法 目的:方法确认——1、加标三组 回收率80~105之间适用范围:饮用水/水源水中的三氯甲烷、四氯化碳 2、精密度六组 RSD≤7.2%仪器:安捷伦气相7820A,柱子:HP-5 3、双平行样 用是超纯水固未检出进样方法:手动进样 (100 μl进样针) 4、质控样(方法确认时质控样未到货,固是本实验的遗憾后面会验证)进样体积:30 μl 疑问:从色谱图中可以看出,在目标物三氯甲烷/四氯化碳前都会出现一个很大的峰,不能确定标准品:购买的三氯甲烷、四氯化碳的储备液2000 mg/L 这是什么造成的,想咨询各位老师,给我进行解答。(因为是气体手动进样,固不可能 是溶剂峰。)标曲范围:三氯甲烷—0、0.2、1、2、4、10 μg/L 解答:如果大家有什么想问的或者咨询的可以一起讨论。 四氯化碳—0、0.1、0.5、1、2、5 μg/L定量方法:峰面积http://ng1.17img.cn/bbsfiles/images/2016/12/201612071515_01_2867868_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612071515_02_2867868_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612071518_01_2867868_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612071518_02_2867868_3.png
请教各位谁做过纯净水中的四氯化碳和三氯甲烷吗?用气相测的条件是什么?要什么需要注意的?
用的是Thermo 的GC色谱仪按照国标GBT 5750.8 方法做,从同一瓶三氯甲烷四氯化碳混标出来的标准系列5个点(0.2—5 ppb),线性为,0.91;达不到0.99孵化池40℃平衡了1小时,进样量0.3mL,色谱条件和标准一样,出来的峰型很好顶空瓶120℃加热了2h,密封垫圈也用煮沸的水洗过晾干衬管换下来清洗过,柱子应该是没问题的milli-Q超纯水机出来的水检出很高的三氯甲烷四氯化碳响应,故换用了市售屈臣氏蒸馏水(低检出),煮沸的水四氯化碳响应反而变高怎么才能把这个标曲做出来?已经弄了4次了,最好一次是0.91,最坏一次完全没线性。而且空白略高
各位: 我现在手上有个标定三氯化铁的单页标准,先配三氯化铁溶液,然后用硫代硫酸钠标定,具体过程是这样:取15ml三氯化铁溶液,加硫酸1ml,加高锰酸钾试液至粉红色,再加盐酸15ml,碘化钾试液15ml,暗处放置5分钟后滴定,并用空白校正。我现在的问题是,滴定的平行性不好,空白从1.1,1.5,1.7,2.0,样品消耗的滴定液体积也是越来越高。不知道是什么原因。我考虑是不是放置时间有关系,因为我没有严格按照这个来操作。请各位指教!谢谢!
仪器:岛津2010PLUS,ECD检测器、Dani86.50顶空自动进样器色谱条件:色谱柱:RTX-1 30M*0.25mm*0.25um 柱温:60℃ 5min 进样口温度:200℃,检测器:250℃ 分流比:30 柱流速:1.5ml/min顶空条件:平衡温度:50℃,平衡时间:25min,六通阀温度:100℃,传输线温度:100℃今天做水中三氯甲烷、四氯化碳时,标准系列:三氯甲烷(0.02、0.05、0.1、0.15、0.2、0.5和四氯化碳(0.002、0.005、0.01、0.015、0.02),三氯甲烷和四氯化碳都在第四个浓度时色谱峰出现分叉。仪器是新的,色谱柱也是新的。







 我要推广仪器
我要推广仪器
 下载APP
下载APP