请问有没有做过环草定原料环戊酮-2-羧酸乙酯含量的老师,请问用的什么柱子,请赐教!我用的是XE60毛细管柱,但是出峰不好.
增效联磺片为磺胺类抗菌消炎药的新型复方制剂,每片含磺胺甲基异(口恶)唑200mg、磺胺嘧啶200 mg、甲氧苄氨嘧啶80 mg,各地方标准均有收载,对前两种成分以纸色谱法鉴别,而对甲氧苄氨嘧啶则另行鉴别。本文以薄层色谱法同时鉴别磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶3种成分,专一性强,斑点明显,操作简便,结果较为满意。1 仪器与试药 三用紫外线分析仪(上海顾村电光仪器厂),硅胶GF254薄层板(10cm×20 cm,自制);磺胺甲基异(口恶)唑、磺胺嘧啶和甲氧苄氨嘧啶对照品(中国药品生物制品检定所);增效联磺片(市售品);硅胶GF254(青岛海洋化工厂生产,化学纯);其它试剂均为分析纯。2 溶液的配制2.1 单一对照品溶液 分别精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液分别制成0.5 mg/mL磺胺甲基异(口恶)唑、0.5 mg/mL磺胺嘧啶、0.2 mg/mL甲氧苄氨嘧啶的单一对照品溶液。2.2 混合对照品溶液 精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液制成? mL含磺胺甲基异(口恶)唑0.5 mg、磺胺嘧啶0.5 mg和甲氧苄氨嘧啶0.2mg的混合对照品溶液。2.3 样品溶液的配制 取供试品细粉适量(约相当于磺胺甲基异(口恶)唑50mg),加50 %丙酮溶液100 mL,振摇使溶解,过滤,滤液作为供试品溶液。
[table=100%][tr][td]我做的一种羧酸酯液相色谱峰拖尾,流动相是乙腈的反相梯度洗脱,柱子是C8柱,怎么从改变流动相入手消除拖尾呢?[/td][/tr][/table]
请问同志们,羧酸类物质能检测吗??能用甲醇做流动相吗?羧酸会不会和甲醇生成酯,而影响测定羧酸的PH=3-4,需要配制流动相+怎么样的缓冲溶液??????大家帮忙啊
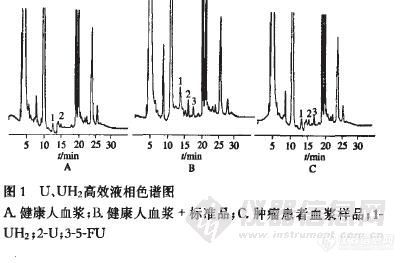
作者:肖力 任斌 陈小陆 李瑞明 刘怡 容颖慈 蓝缨(作者单位:中山大学附属第一医院药学部,广东,广州,510080 )摘要:目的:建立准确测定内源性尿嘧啶和二氢尿嘧啶血药浓度的高效液相色谱法.方法:以氟尿嘧啶(5-FU)为内标,醋酸乙酯-异丙醇混合液(85:15)为提取溶剂;色谱柱Diamonsil C18柱(250 mm×4.6 mm,5 靘);流动相A-0.01 mol·L-1磷酸二氢钾缓冲液(pH 5.5),B-乙腈,梯度洗脱;流速为0.8 mL·min-1;柱温为4 ℃;检测波长为204 nm(0~14.5 min),254 nm(14.5~35 min).结果:尿嘧啶和二氢尿嘧啶线性范围为8~500 靏稬-1,线性回归方程分别为C(UH2)=61.760 8Y+0.506 5,r=0.999 8;C(U)=95.201 1Y-3.064 0,r=0.999 3,(n=7).最低检测质量浓度均为5 靏稬-1.尿嘧啶方法回收率为99.3%~107.0%,二氢尿嘧啶方法回收率为95.0%~98.3%.尿嘧啶日内RSD小于6.5%,日阍RSD小于11.7%,二氢尿嘧啶目内RSD小于9.2%,日间RSD小于12.4%.结论:本方法可用于内源性尿嘧啶和二氢尿嘧啶血药浓度的常规监测.谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208142006_383845_1609970_3.jpg
[color=#444444]请问各位大侠,羧酸和羧酸盐在同一液相色谱条件下出峰的时间是否一样?例如醋酸和醋酸钠,流动相为乙腈和水,PH为酸性,磷酸做缓冲液。另外,羧酸和羧酸盐的极性是否相同?[/color]
[font=Verdana]羧酸吸收什么情况下会消失?[/font]除了重水交换以外,还有别的情况会导致羧酸上的H的核磁吸收消失吗?
[color=#444444]最近用安捷伦6495[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]想着对尿嘧啶摸方法,TIC图里面母离子不出峰,而且在最开始的时候有倒峰,靠近最后的时候有一个149的不知知道是什么的峰,求大神解答[/color]
我最近买了进口的标品,买的时候说是磺胺嘧啶,结果说明上的英文是SULFANILAMIDE这个单词是磺胺的意思。请问一下这两个是一个东西么?
根据欧盟委员会(EC)No 396/2005法规第6节的规定,澳大利亚收到一份来自AGRIPHAR S.A公司要求欧盟修改莴苣和菊苣中乙胺嘧啶(pyrimethanil)杀虫剂的最高残留限量(MRL)的申请。为了与欧洲南部和北部的气候相适应,澳大利亚决定提高这些作物中乙胺嘧啶的最大残留限量(MRL)。澳大利亚依据欧盟委员会(EC)No 396/2005法规第8节的规定起草了评估报告,并提交至欧盟委员会,之后于2010年12月1日转至欧洲食品安全局。欧洲食品安全局对澳大利亚根据91/414/EEC指令提交的评估报告草案(DAR)进行了审核,对乙胺嘧啶的毒理学概况进行了评审,认为新的MRL符合其0.17 mg/kg bw/d的ADI值,并不会对消费者构成公众健康风险,做出如下决定:代码商品现有的最大残留限量 (毫克/千克)提议的最大残留限量 (毫克/千克)对提议的建议执行的残留物质:乙胺嘧啶0251020莴苣1020拟议的MRL残留量数据充分,不会对消费者构成健康风险。0251030菊苣1020
最近在做一个建立岛津高效液相色谱法测定5-氟尿嘧啶浓度的试验,用5-嗅尿嘧啶做内标,但是5-氟尿嘧啶和5-嗅尿嘧啶没有分离开,应该怎么办?
作者:何冰题目:5-氟尿嘧啶壳聚糖微球的制备与表征期刊:天津大学年份:2007链接:http://www.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&QueryID=4&CurRec=186&dbname=CMFD0911&filename=2008186937.nh&urlid=&yx=
要测葡萄糖中C2-C5的一元羧酸检测方法请高手执教!
请问大家:嘧啶pyridine可以在GC-FID上run吗?如果可以,应该是非极性或弱极性吧,但是为什么可以和水互溶呢?另外,嘧啶在衍生化过程中的作用是什么呢?thx
为什么磺胺嘧啶跑出两个峰,基线也不稳[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2021/01/202101071948511820_5402_5077097_3.png[/img]
减水剂是一种重要的混凝土外加剂,能够最大限度地降低混凝土水灰比,提高混凝土的强度和耐久性。减水剂分为普通减水剂和高效减水剂,减水率大于5%小于10%的减水剂称为普通减水剂,如松香酸钠、木质素磺酸钠和硬脂酸皂等;减水率大于10%的减水剂称为高效减水剂,如三聚氰胺系、萘系、氨基磺酸系、改性木质素磺酸系和聚羧酸系等。在众多高效减水剂中,具有梳形分子结构的聚羧酸系高效减水剂因其减水率高、坍落度保持性能良好、掺量低、不引起明显缓凝等优异性能,成为近年来国内外研究和开发的重点。 在国外,聚羧酸类减水剂的研究已有相当长的历史,其应用技术已经成熟。日本是研究和使用聚羧酸类减水剂最多也是最成功的国家,1995年以后聚羧酸系减水剂在日本的使用量就超过了传统的萘系减水剂,1998年底聚羧酸系减水剂产品已占所有高性能AE减水剂产品总数的60%以上,其主要生产厂商有花王、竹本油脂、日本制纸、藤泽药品等[1]。对聚羧酸系减水剂的研究主要集中在新拌混凝土有关性能和硬化混凝土的力学性能及高强高性能混凝土在工程中的应用技术。目前聚羧酸系减水剂可使混凝土的水灰比下降到0.25以下,而水泥用量仍可保持在500 kg/m3,同时它的坍落度可保持200 mm以上,完全满足施工要求。近年来,北美和欧洲的一些研究者的论文中也有许多关于研究开发具有优越性能的聚羧酸系减水剂的报道,主要是商业开发和推广,如Grance公司的Adva系列、MBT公司的pheomixTOOFC牌号、Sika公司的Viscocrete3010等。 由于成本和技术性能问题,国内对聚羧酸类减水剂产品的研究仅处于实验室研制阶段,只有少量用作坍落度损失控制剂与萘系减水剂复合使用。而且可供合成聚羧酸类减水剂的原料也极为有限,国内原材料单甲氧基聚乙二醇(MPEG)供应不足,MPEG国内没有商业化,必须依靠进口,也有研究人员[3]用聚乙二醇(PEG)代替MPEG,但是由于在制备过程中,双官能度的PEG容易产生交联,使得产品性能较差,质量不稳定。可以说,从减水剂原料到生产工艺、降低成本、提高性能等许多方面都仅仅是处于刚起步阶段。 [color=#DC143C]本文主要对聚羧酸系高效减水剂的化学结构、主要作用机理、合成方法及结构与性能的关系进行了综述。[/color]
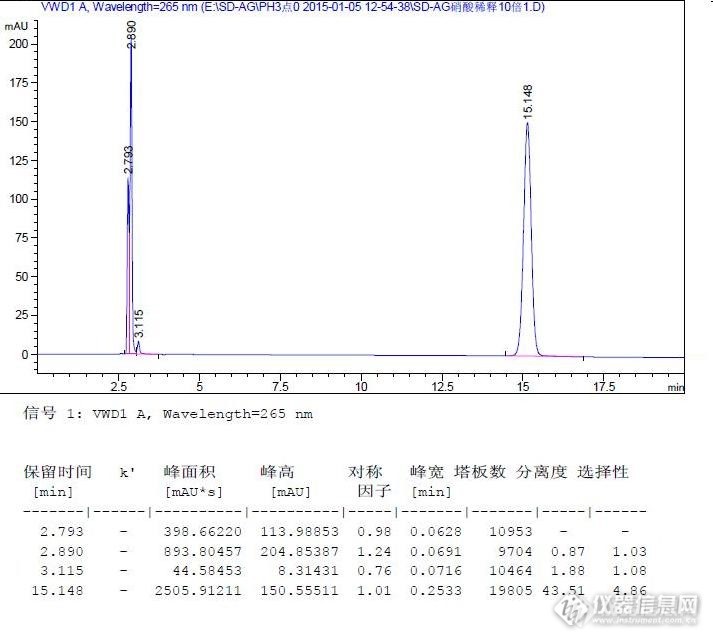
摘要:优化筛选磺胺嘧啶银的含量测定方法。考察了流动相中pH对磺胺嘧啶银保留时间的印象,并与文献中的报道进行了对比,结果表明,月旭的色谱柱,确实值得深度信任。关键词:高效液相色谱法、流动相、pH、磺胺嘧啶银 前言:磺胺嘧啶银,Silver sulfadiazine或silvadene,是一种磺胺类/银盐抗细菌药,化学式为C10H9AgN4O2S,为白色或类白色的结晶性粉末,遇光或遇热易变质。用于治疗烧烫伤创面感染,除控制感染外,还可促使创面干燥、结痂和促进愈合。涂药后,遇光渐变成深棕色。http://ng1.17img.cn/bbsfiles/images/2015/01/201501071447_531476_1609327_3.jpg该品种在2010年版中国药典二部中有收载,其中的含量测定方法为硫氰酸铵滴定法。有关物质研究为TLC法。为了增加对该品种的研究深度,查找文献开发该目标物的液相检测方法。1.实验部分2.设备和试剂高效液相色谱仪,Agilent1260;VWD检测器。乙腈、磷酸均为色谱纯,浓氨溶液、硝酸均为分析纯。1.2色谱条件检测波长:265nm;进样量:10μL;流动相:0.1%磷酸-乙腈(90:10)为流动相,等度洗脱;色谱柱:月旭,Ultimate®,XB-C18,4.6*250mm,5μm(Part. No:00201-31043,Seri. No:211303968),pH适用范围为1.5~10.0。1.3 溶液配制磺胺嘧啶银供试液配制:取本品10mg,精密称定,用20%氨水溶液溶解并定容至10ml。精密量取1.0ml,用10%硝酸溶液稀释并定容至10ml。流动相配制:因所用硝酸的质量分数为85%,取硝酸1.18ml加入到1000ml水中。经pH计测定pH为2.1。分取0.1%的硝酸溶液各250ml两份,其中一份用氨水调节pH至3.0,另一份用磷酸调整pH至1.5。然后按比例配制成三种不同pH的流动相。3.结果与讨论3.1典型色谱图 pH=1.5色谱图http://ng1.17img.cn/bbsfiles/images/2015/01/201501071448_531477_1609327_3.jpg pH=2.1色谱图http://ng1.17img.cn/bbsfiles/images/2015/01/201501071448_531478_1609327_3.jpg pH=3.0色谱图http://ng1.17img.cn/bbsfiles/images/2015/01/201501071455_531481_1609327_3.jpg3.2结果与讨论①. 月旭的色谱柱,检验该品种的峰型良好、理论塔板数高。②. 随着pH的增加,保留时间延长。但理论塔板数并没有随着保留时间的延长而降低。③. 文献中提到,流动相的pH对主成分峰的峰型有较大影响。而本实验表明,pH在2~3的范围内,峰型都良好。表明对流动相的耐用性良好。 http://ng1.17img.cn/bbsfiles/images/2015/01/201501071448_531480_1609327_3.jpg
我想做三羧酸循环中a-酮戊二酸标记或乙酰辅酶A中碳的同位素标记然后研究三羧酸循环和这种植物所形成的色素的关系,可是我没有同位素标记设计的经验,谁可以帮帮我,谢谢啦~~~
吡啶二羧酸酐结构看似简单,可因其在水中水解成酸,影响样品游离酸含量分析,化学滴定不好作;做GC时热分解,与相应的二酸峰重合;液相用水溶液作流动相不行,用非水有机相作流动相如用醇又会醇解,估计可用正相做,但该样在许多有机溶剂中溶解性差,所以一直没找到好的分析方法。请问哪位老师作过吡啶二羧酸酐的HPLC分析,能否帮帮我。谢放!
哪位大神 帮我查下酒石酸噻嘧啶的红外图谱 英文名:Pyrantel Tartrate ...................................................
氟甲阿糖尿嘧啶(又稱作L-FMAU) 為一種藥物其為核苷類似物其有4種異構物L-form的FMAU和D-FMAU 其中L和D都各有α/β共有4種L的α/β和D的α/β圖譜上有何不同~~請高手指教
我看的文献方法衍生全氟羧酸,用三乙基硅烷醇的方法,用的仪器是岛津的单杆EI 源,但是衍生以后全扫模式下,所有的全氟羧酸出的峰都一样。通过SIM模式下才能找到目标峰,并且PFDA/PFNA/PFDOA的峰都非常小。我用的是1ug/ml得标液衍生的,全氟辛酸的峰大概只有1000,其他的峰高就只有100不到。有没有大神做过类似的方面,求帮助。还有一个问题,如果做全氟羧酸的目标物,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]做的话,文献中有用NCI源和EI源的,具体的那个方法更好一点呢。跪谢!
我想分析甲酰嘧啶和氨基嘧啶的混合物,不知道用什么流动相?用的HPLC(紫外检测器),用紫外分光光度计扫描过了,氨基嘧啶和甲酰嘧啶的最大吸收波长都在274 nm,试过几种流动相,两者根本分不开。
HJ 1220-2021 6种羧酸类的测定 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱法 一般的柱子的可以分析吗
工作场所羧酸类,我只有ffap极性毛细管柱,方法中乙酸是用甲酸作为溶剂,可是甲酸腐蚀性太大,不敢做,有没有其它溶剂能代替的,还有氯乙酸响应实在太小,峰型很难看,检出限远达不到国标,是色谱柱的问题吗?哪位同行做过,详解一下
据欧盟食品安全局消息,鉴于农药甲基嘧啶磷(pirimiphos-methyl)的交叉污染风险,11月15日欧盟食品安全局对甲基嘧啶磷的最大残留限量进行了审查,经审查欧盟食品安全局建议对甲基嘧啶磷在多种作物中的最大残留限量进行修订。 欧盟食品安全局在风险评估过后,做出如下决定:商品代码商品现行MRL(mg/kg)建议MRL(mg/kg)401000油料作物种子0.050.5500010大麦55500020荞麦50.5500030玉米50.5500040小米55500050燕麦55500060大米50.5500070黑麦50.5500080高粱55500090小麦(包括黑小麦)55810000调味料(种子)55820000调味料(果实与浆果)0.10.1830000其它的植物商品见附录B0.051000000动物源食品0.050.01 原文链接:http://www.efsa.europa.eu/en/efsajournal/doc/2436.pdf
测试C18色谱柱的柱效,用到尿嘧啶,主要是看该色谱柱的死时间大小。由于尿嘧啶出峰时间短,理论塔板数也相应较小。近日遇到这样的投诉,甲苯的塔板数可以达到证书上的85%,但是客户觉得尿嘧啶的塔板数过小,为难的是厂家的证书上连尿嘧啶的塔板数都没有,那么尿嘧啶的塔板数是否具有参考的价值呢
求用羧酸合成酰 氯,羧酸分子350以上,酰 氯如何分析,酰 氯反应如何中控?
1.C…O键强:介于C=O与C―O之间,强偶合。 2.光谱特征 不对称伸缩谱带:1650~1550 cm?1,显著。 对称伸缩谱带:1400 cm?1附近,较弱。 3.结构确证方法 转化为盐:羧酸与脂肪族叔胺(如三乙胺)在氯仿中反应(四氯化碳无效)。 4.特征谱带 羧酸根离子:两个羰基吸收谱带。 “铵”谱带:2700~2200 cm?1。 O—H伸缩谱带:消失。
1.O-H伸缩振动羧酸在浓溶液或固态中因强氢键成二聚体。 强氢键源于离子共振,阻碍游离羟基振动,仅稀非极性溶剂或蒸气相中可见(约3520 cm?1)。 二聚体O-H振动宽且强,范围3300~2500 cm?1,常集中于3000 cm?1,伴弱C-H振动。 长波长精细结构为倍频与复合频。 β-二酮等也有此吸收,但较弱,C-O振动频率较低。与醚类溶剂形成分子间氢键,O-H吸收约3100 cm?1。 2.C-O伸缩振动羧酸C-O振动强于酮,单体约1760 cm?1。二聚体对称,仅不对称振动有吸收,氢键与共振降低频率至1720~1706 cm?1。 分子内氢键影响更大,如水杨酸1665 cm?1,对羟基苯甲酸1680 cm?1。 不饱和共轭轻微降低频率,α,β-不饱和及芳基共轭酸二聚体约1710~1680 cm?1。 α位电负性取代基(如卤素)轻微增加频率,旋转异构致双重谱带。 3.C-O伸缩与O-H弯曲振动羧酸红外光谱中,C-O伸缩约13201210 cm?1,O-H弯曲约14401395 cm?1,两者有相互作用。 二聚体C-O伸缩强吸收约1315~1280 cm?1,长链脂肪酸呈双峰。 O-H面外弯曲特征谱带约920 cm?1,中等强度峰宽。







 我要推广仪器
我要推广仪器
 下载APP
下载APP