各位兄弟姐妹们: 有谁了解聚蓖麻油酸、四聚蓖麻油酸,他们的是怎么产生的,有什么区别等等,越详细越好!先谢谢啦!
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
如题:蓖麻油酸中的苯酚怎么检测含量?如果直接进入气相色谱,植物油酸的汽化点太高,不宜直接汽化。
蓖麻油方法: GC基质: 植物油应用编号: 101924化合物: 1-棕榈酸甲酯2-硬脂酸甲酯3-油酸甲酯4-亚油酸甲酯5-蓖麻油酸甲酯固定相: DM-FFAP色谱柱/前处理小柱: DM-FFAP 30m x 0.32mm x 0.5u商品编号: 7633 样品前处理: 取蓖麻油40mg,置50mL圆底烧瓶中,加入0.5mol/L氢氧化钠-甲醇溶液5mL,在60℃水浴中回流30min,至油滴全部消失,再加入三氟化硼乙醚-甲醇(1 : 3, v/v) 4mL,回流5min,冷却,精密加入正己烷5mL,振摇5min,分取正己烷,用饱和氯化钠溶液洗涤两次,每次5mL,放置,取上清液,置10mL具塞试管中,加1g 无水硫酸钠脱水,振摇,精密量取上清液1mL,置10mL量瓶中,用正己烷稀释至刻度,摇匀,即得。 色谱条件: 色谱柱: FFAP (柱长为30.0m,内径为0.32mm,膜厚度为0. 5μm)流动相: 高纯氮流速: 1.0 mL/min柱温: 200℃(11min) 5/min 240℃(10min)检测器: FID进样量: 1μL 关键字 :蓖麻油;1-棕榈酸甲酯;硬脂酸甲酯;油酸甲酯;-亚油酸甲酯;蓖麻油酸甲酯;gc;DM-FFAPhttp://www.dikma.com.cn/Public/Uploads/images/213(1).JPG峰号保留时间min峰面积μV*s峰高μV理论塔板数NUSP拖尾因子分离度18.190186.332.4467420.982212.947160.820.5627361.02313.582478.961.9740250.96414.806672.191.6945571.06528.3251596180.63126230.98备注说明1- 棕榈酸甲酯2- 硬脂酸甲酯3- 油酸甲酯4- 亚油酸甲酯5-[font='Tim
有人做过2015版中国药典的氢化蓖麻油脂肪酸组成吗?为什么会12羟基硬脂酸甲酯偏低呢?前处理的回流萃取,哪一步需要注意呢?
求助,哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做确认,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰面积峰形都相近,RSD很小,但是随行的对照品溶液在进了6针样品后峰形变矮且拖尾,峰面积也差了好几倍,有做过的这样的吗?
求蓖麻油含量测定,供试品含量公式,还有大神们怎么制备的供试品的,为啥我做出来的含量只有20%左右
有哪位大神做过2015版中国药典的氢化蓖麻油脂肪酸组成呢?为什么我做出来俩结果不平行 两针对比下来有一针12羟基硬脂酸甲酯偏低 而硬脂酸甲酯偏高 难道二者之间存在转化关系么?还有实验过程中有什么需要注意的吗?
网上查,蓖麻油的分子量很大 不知道用气质能否检测是否出峰?
公司新进的蓖麻油测定时发现按药典方法上面用正己烷5毫升提取一次不能完全提出来,含量只有百分之十几,不知道大家都是怎么操作的[img]https://ng1.17img.cn/bbsfiles/images/2019/12/201912100733389029_7043_3396781_3.png[/img]
如题:哪位版友有蓖麻油的具体检测方法及标准?盼应助!
那位大虾知道蓖麻油的质量指标是什么?请赐教
求助,有哪位大神做过2020版中国药典的氢化蓖麻油脂肪酸组成呢?最近在做这个,配制的对照品溶液连续进样6针,12-羟基硬脂酸甲酯峰峰形和峰面积都很相近,但是在进样6针样品后的随行对照品溶液,12-羟基硬脂酸甲酯峰的峰形变矮且拖尾,峰面积也变小了一半多,更换了衬管后刚开始峰面积正常,进了样品后就不行了,有什么解决方法吗
蓖麻油样品 测试项目:含量测定 样品配制: 取蓖麻油40mg,置50mL圆底烧瓶中,加入0.5mol/L氢氧化钠-甲醇溶液5mL,在60℃水浴中回流30min,至油滴全部消失,再加入三氟化硼乙醚-甲醇(1 : 3, v/v) 4mL,回流5min,冷却,精密加入正己烷5mL,振摇5min,分取正己烷,用饱和氯化钠溶液洗涤两次,每次5mL,放置,取上清液,置10mL具塞试管中,加1g 无水硫酸钠脱水,振摇,精密量取上清液1mL,置10mL量瓶中,用正己烷稀释至刻度,摇匀,即得。 仪器型号:Agilent 6890N分析条件 色谱柱: FFAP (柱长为30.0m,内径为0.32mm,膜厚度为0. 5μm) 流动相: 高纯氮 流速: 1.0 mL/min 柱温: 200℃(11min) 5/min 240℃(10min) 检测器: FID 进样量: 1μL 实验谱图http://ng1.17img.cn/bbsfiles/images/2012/07/201207161029_377774_2370618_3.jpg 实验数据 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 1 8.190 186.3 32.4 46742 0.982 2 12.947 160.8 20.5 62736 1.02 3 13.582 478.9 61.9 74025 0.96 4 14.806 672.1[
药典蓖麻油含量测定 供试品制备里,如图绿色部分应该是在氢氧化钾的催化下和甲醇进行酯交换反应,但如图红色部分加三氟化硼乙醚-甲醇(1:3)是什么目的?求指教
最近在做氢化蓖麻油脂肪酸组成,按药典方法做 ,每次都是12-羟基硬脂酸出峰偏低,感觉没有衍生化完全,各位大神能告知一下有什么注意点吗。我每次做出来其他峰面积都差不多,就是12-羟基硬脂酸的峰面积有差异,而且以面积归一法计含量也只有70%到不了要求的78-91%。
大家好!小弟是做化妆品原料检测的,在测量氢化蓖麻油(12-羟基-十八烷酸甘油酯)的碘值时,溶剂采用氯仿,测出的结果比供应商COA文件给出的大,但供应商说是方法不同,他们的溶剂用的CCl4,并且加了乙酸汞,测出的结果正好在范围之内,我想问用这两种溶剂测出的碘值有啥不同?还有乙酸汞的加入是不是只起到缩短检测时间的作用,对最终的结果并无影响??
各位,请问氢化蓖麻油脂肪酸组成检测硬脂酸含量偏高,12-羟基硬脂酸偏低为什么呀
最近在做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]氢化蓖麻油脂肪酸组成检验,有做过的吗?
最近小弟按照欧盟的标准做氢化蓖麻油的镍的限度,限度是不超过1ppm,可是厂商的报告是0.1ppm,并且我做的结果老是忽高忽低,第一天测的是1.2ppm,第二天测得是1.3ppm,第三天测得1.5ppm,第四天测得是0.15ppm,同时第四天的样品空白值比前几天高很多,也测了以前合格的样品,可检测结果又高出了限度值。很是不解。我是用石墨炉法,微波消解!请教各位大侠,在配制、消解和测量的的过程中,要注意哪些细节啊?非常感谢!!
我想查询一些化工原料的质保时间和存储要求。具体有下列原料在:固体:苯酐 环氧树脂 新戊二醇 三羟甲基丙烷 间苯 偏苯 己二酸 苯甲酸 过氧化苯甲酰液体:甲甲酯 丙烯酸 丙烯酸丁酯 甲基丙烯酸 苯乙烯 丙烯酸羟乙酯 丁醇 乙醇 单丁醚 丙二醇甲醚 过氧化苯甲酸叔丁酯 二甲基乙醇胺 豆油酸 脱水蓖麻油酸 亚麻仁油酸。
有没有人做ELSD检测脂肪酸酯,聚甘油酯,聚甘油,蓖麻油酸酯等等,有的话是否可以分享一下或交流一下
我用的是 油酸 蓖麻油 橄榄油 IPM 请问 怎么测药物在它们中的溶解度
可以从红外上区分醇酸树脂是蓖麻油、亚麻油、桐油或大豆油4者中的某一个合成的吗?谢谢!
[color=#444444]最近在研究一种药物的液相色谱分析方法,药品说明书上是这样写的:[/color][color=#444444]本品每粒含多烯磷脂酰胆碱(天然多烯磷脂酰胆碱,带有大量的不饱和脂肪酸基,主要为亚油酸(约占70%)、亚麻酸和油酸)228mg。辅料为:氢化蓖麻油,硬脂肪、乙基香兰素,4-甲氧基苯乙酮、乙醇、空心胶囊。[/color][color=#444444]磷脂酰胆碱是药效成分,胶囊里面还有其它少量的成分(不是辅料)。[/color][color=#444444]有两个问题:(1)它说的228mg是胶囊里面所有物质的质量(辅料除外),不是磷脂酰胆碱的质量?[/color][color=#444444](2)如果228mg是包含了所有物质,那我做磷脂酰胆碱的含量测定,结果测出来了有用吗?比如用外标法测出的结果是670mg/g。因为说明书里没有要求磷脂酰胆碱是多少,只给了个总质量,那我测的结果怎么判断是否符合它的要求?[/color]
蓖麻油甘油单酯液相检测的方法?样品:0.1g含蓖麻油单甘油酯的样品溶于2ml异丙醇中。流动相:异丙醇:乙腈(体积比1:1)色谱柱:c18反相柱。但最终还是检测不出来,望各位为小弟指点迷津!谢谢!
花生油,大豆油,蓖麻油,加上婴儿油,瞬间感觉好诡异。为啥有的按来源有的按用途呢?
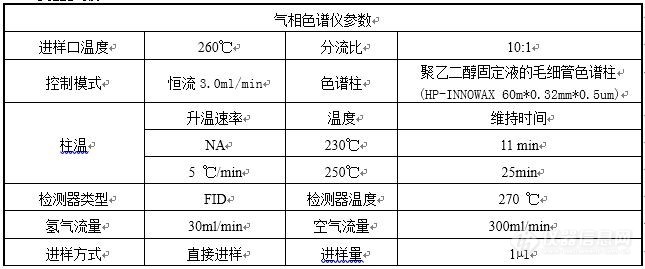
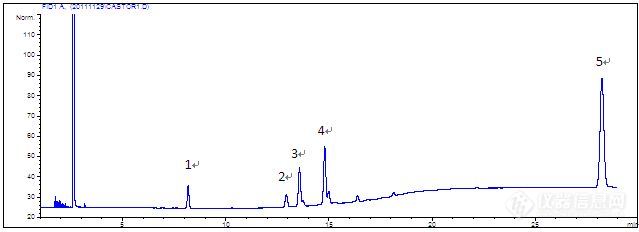
一、方法来源:2020年版《中国药典》二、色谱条件:[img=色谱条件,645,269]https://ng1.17img.cn/bbsfiles/images/2022/03/202203091537079436_3314_3109800_3.jpg!w645x269.jpg[/img]三、描述:1、药典对色谱柱要求只有“聚乙二醇”,但考虑最高柱温有250℃,所以选择了INNOWAX。同时发现30m柱子分不开12-氧基硬脂酸甲酯和12-羟基硬脂酸甲酯,所有选择了60m柱子。2、系统适用性溶液图谱正常,5个已知成分完全分离(药典采集时间只有25min,此处延长到40min)[img=SYS,690,240]https://ng1.17img.cn/bbsfiles/images/2022/03/202203091546319907_3937_3109800_3.jpg!w690x240.jpg[/img]3、供试品第一针图谱亦正常[img=,590,210]https://ng1.17img.cn/bbsfiles/images/2022/03/202203091552128384_7400_3109800_3.jpg!w590x210.jpg[/img]4、重复性实验时,连续6针供试品溶液图谱中,主峰(12-羟基硬脂酸甲酯)拖尾越来越严重[img=,472,575]https://ng1.17img.cn/bbsfiles/images/2022/03/202203091554134141_894_3109800_3.jpg!w472x575.jpg[/img]四、已调查过的影响因素1、最开始进样口的分流比为1:1,系统适用性溶液图谱正常,但是所有的供试品溶液图谱中主峰均拖尾。分析后认为是供试品溶液过载,所以才调整为10:1。但是除了第一针正常,其他都拖尾。后来调整为20:1还是拖尾。2、衬管污染,更换新的超高惰性衬管后,供试品溶液主峰依然拖尾。3、色谱柱污染,截弃色谱柱前段约30cm,老化后测试,供试品溶液主峰依然拖尾。以上问题,希望各位老师能指点指点!谢谢!
前几天在论坛上贴了个我们老师自制的香皂,大家都问用什么方法做的,今天拿来和大家分享一下,希望大家喜欢实验室个人香皂制作方法制作香皂的时候所使用的皂基是棕榈油和蓖麻油,蓖麻油的作用主要是增加香皂的透明感,但是我们自己做出的透明效果不是很好,这个主要看个人喜好。棕榈油的主要作用就是增加香皂的坚硬性,整个过程时间比较长,因为是全手工做的,没有任何可以增加产率的机械设备。并且效果也确实没有专业生产出来的好,但自己做出来的东西,自己心里明白,效果怎么样自己可以调节,作重要的是使用安全放心。主要配方:150g 的水73.5g 的碱液250g 的棕榈油100g 的椰子油150g 的蓖麻油酒精混合液:195g 的酒精105g 的甘油75g 的砂糖(与50g 的温水)主要原料:油脂类:棕榈油,椰子油,蓖麻油,(或)硬脂酸碱液酒精混合液:乙醇,甘油,砂糖冷却反应设备其他:反应防蒸发设备,搅拌等基本工具制作方法:1、在温度45℃-55℃(这个可以自己调节)混合水、油脂、碱液搅拌到美乃滋状。2、加如甘油,然后加热到皂基变为胶状后即可,然后加入酒精3、然后继续搅拌,如果有 凝固为固态的,用汤匙压平混合。4、大概十分中后加入砂糖混合液5、等皂基形成糖浆状之后,加入精油和其他添加物。6、去掉硬块或者泡沫7、全部弄出来之后放入提前准备好的模型里面,等皂基变硬后拿出来切成自己想要的大小块,(切记不要让香皂太硬了,要不然就不好切了)8、切好之后就放在干净的地方让他干燥了,差不多得两三个星期就差不多了注意事项:1、制作过程中不能有残留的油脂,所以,不能减少碱液2、皂基含量太多,透明效果会很不好,颜色也不好。砂糖或酒精放多了的话香皂会软趴趴的,黏黏的。所以原料比例要掌握好。3、干燥过程中要将水和酒精蒸发干净。4、最后经过使用之后发现自己做的香皂比起普通的香皂来说比较软,碰到水之后比较容易变软,我一般用完之后会记得及时晾干。5、其他安全措施自己注意就是了 终于写完了,话说快中午了还没吃早餐,先祝各位周末快乐生活中有好多东西我们完全可以自己做,虽说效果不是很好,玩至少用着安全放心,如果大家还有什么其他好的东西都一起来分享分享,吼吼 还有还多话要说………………







 我要推广仪器
我要推广仪器
 下载APP
下载APP