请教:买来的液体的愈创木酚怎么配置0.05mol/L的溶液,100mL?是用95%的乙醇配吗?
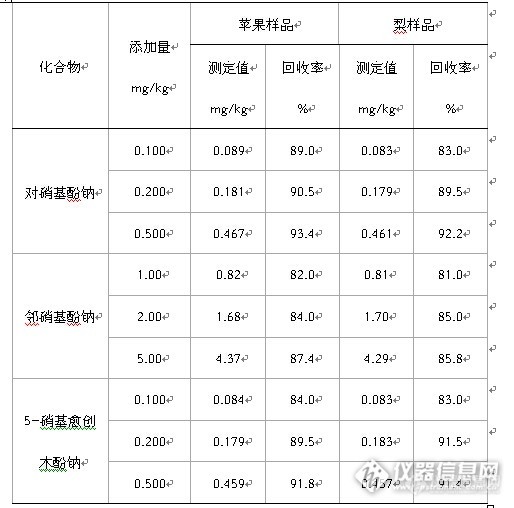
UPLC-MS/MS法测定水果中复硝酚钠残留量本文通过优化质谱参数和色谱条件,考察流动相、提取方法等因素,建立了UPLC—MS/MS测定水果中复硝酚钠残留的分析方法。该方法操作简单,快速,方法灵敏度、回收率及重现性均令人满意。1 材料与方法1.1 材料与试剂对硝基酚钠,邻硝基酚钠和5-硝基愈创木酚钠标准品纯度≥98%;乙腈,甲醇为色谱纯;实验室用水为超纯水;所用试剂除特殊说明外均为分析纯。1.2 仪器UPLC-MS/MS 超高效液相色谱串联质谱仪(QuattroPremier XE) 美国沃特世公司,配电喷雾离子源;电子分析天平 梅特勒-托利多仪器(上海)有限公司; 1.3 标准溶液的配制准确称取对硝基酚钠,邻硝基酚钠和5-硝基愈创木酚钠标准品(精确至0.1mg),分别用水溶解制得标准储备液。取各标准储备液适量,用水稀释制得混合标准工作溶液,其中对硝基酚钠、5-硝基愈创木酚钠浓度配制成0.00,0.05,0.10,0.20,0.50,1.00,2.00mg/L,邻硝基酚钠配制成0.00,2.5,5.0,10.0,25.0,50.0,100.0mg/L。该溶液于4℃冰箱内保存。1.4 样品处理称取已匀浆的样品5 g (精确到0.0001 g),放入具塞150mL碘量瓶内,加人甲醇提取液20mL,盖上玻璃塞,超声振荡5min,过滤,滤渣加入10mL甲醇提取液,超声振荡5min,过滤,滤渣加入lOmL甲醇提取液,超声振荡5min,残渣弃去,合并3次提取液,在提取液中加入10g无水硫酸钠脱水,然后用无水硫酸钠过滤,并用20mL甲醇冲洗,收集滤液在旋转蒸发仪上45℃减压蒸发浓缩近干,用水定容至5mL,过0.22μm滤膜,待测。1.5 仪器操作条件1.5.1色谱条件色谱柱:Waters ACQUITY UPLC HSS T3 (4.6 mm ×100mm,1.8 μm);流动相A:甲醇流动相B:10 mmol/L乙酸铵水溶液。等度洗脱程序:流动相A:60% ;流动相B:40% ;进样量:5 μL;流速:0.2 mL/min;柱温:30℃ 。1.5.2质谱参数离子源:电喷雾离子源(ESI),采用ESI- 模式;扫描方式:多反应监测(MRM);毛细管电压(Capillary):3.5kV;锥孔电压(Cone):30V;离子源温度(SourceTemp):110℃;脱溶剂气温度(Desolvation Temp):450℃;脱溶剂气流量:450L/hr;锥孔气流量:50L/hr;碰撞气流量:0.12ml/min;驻留时间:0.2 S。三种物质的保留时间、母离子、子离子、锥孔电压、碰撞电压和驻留时间见表1。三种物质的总离子流图见图1,5-硝基愈创木酚钠总离子流图和定性离子对图见图2,对硝基苯酚钠和邻硝基苯酚钠总离子流图和定性离子对图见图3.表1 三种物质的保留时间、母离子、子离子、锥孔电压、碰撞电压和驻留时间化合物保留时间min母离子(m/z)子离子 (m/z)锥孔电压(eV)碰撞电压(eV)驻留时间(S)5-硝基愈创木酚钠2.21168.15152.9725150.2123.1225250.2对硝基酚钠2.23138.15108.1230150.292.1830250.2邻硝基酚钠2.83138.15108.1230150.2[fo
按EPA8270做环境检测方面的半挥发性有机物,硝基酚和五氯酚等标准物质在MS上不出峰,有高手请指教下。[em01]
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能够将间位硝基苯酚、对位硝基苯酚、邻位硝基苯酚能分开么?谢谢
基本上是按照GB/T 21311 做的 10ml盐酸 0.2M200ul 二硝基苯甲醛 0.1M1PPM标品37℃水浴振荡16h加1ml磷酸钾 用氢氧化钠调至7.4 10ml ETOAC 振荡 离心 氮吹求解为什么找不到母离子是衍生的不好? 还是其他原因
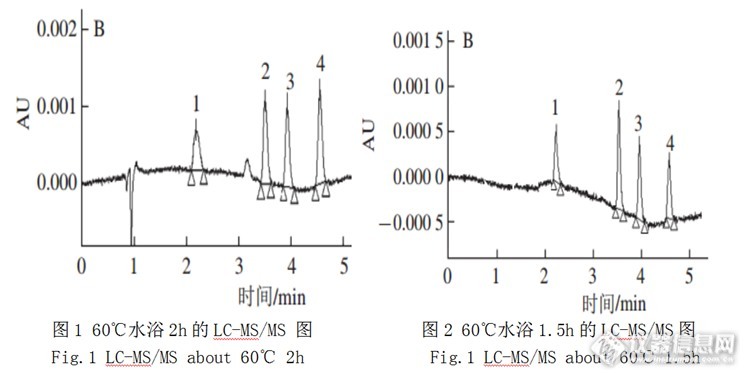
本人对肉粉这种基质复杂的样品进行处理,同时结合硝基呋喃代谢物提取时间长,提取率不高的问题进行优化,最终确定最佳的条件。[align=center][b]肉粉中硝基呋喃代谢物的高速动能前处理方法[/b][/align][align=center]王成梅[/align][align=center](山东中质华检测试检验有限公司,山东省 济宁市 邮编272000;)[/align][b]摘要:[/b]采用高速动能前处理技术,建立了快速、提取率高、回收率好的肉粉中硝基呋喃代谢物的液相色谱-串联质谱方法。处理后的样品添加同位素内标、酸水解、2-硝基苯甲醛衍生化、乙酸乙酯提取,正己烷除油,采用C18色谱柱进行分离,乙腈-0.002mol/L的乙酸铵为流动相进行梯度洗脱,多反应监测(MRM)模式检测。结果表明,采用高速动能处理过的样品在60℃经过2h衍生后,四种硝基呋喃代谢物在各自的线性范围内线性关系良好,相关系数均大于0.9990;方法的检出限在0.2ug/kg;通过三个低、中、高水平的加标,平均回收率(n=3)为91.7-105.2%,标准偏差小于3.10%。本方法采用一种新型的处理样品的方法,缩短了时间,提高了效率,增加了回收率,适合于基质复杂的肉粉中硝基呋喃代谢物的测定。关键词:高速动能;硝基呋喃代谢物; 肉粉; 液相色谱-质谱方法Quantitative Determination of Four Nitrofuran Metabolites in Meat Meal using high speed kinetic energy pretreatment technology[b]Abstract[/b]:Using high speed kinetic energy pretreatment technology, a method for the simultaneous determinatin of four nitrofuran metabolites in meat meal by liquid chromatography tandem mass spectrometry([url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)was eatalished.The method has a rapid ,high extraction and recovery rate.The homogenized sample was spiked with isotope inter standards,hydrolyzed withHCl,derivatized with nitrobenzaldehyde,extracted with ethyl acetate,and degreasing by hexane.The analysis was carried out on C18 column by gradient elution with acetonitrile-ammonium acetate as mobile phase,and detected by MS/MS system with positive electrospray ionization under multiple reaction monitoring(MRM)mode.The compounds was identified with retention time and ion ratio and quantified by the internal standard calibration curve isotope dilution technique.After 60℃ derivation,the results showed that the correlation coefficients(R20.9990)of four targets were obtained within their respective linear ranges over dynamic range of 0.2-5μg/L .The average recoveries were ranged from 91.7%-105.2% for the four targets at three spiked levels with the relative standard derivation (RSD,n=6)were under 3.10%.The method was precise and sensitivity,suitable for quantitative and qualitative analysis of four nitrofuran metabolites in meat meal.[b]Key words[/b]:High-speed kinetic energy nitrofuran metabolites meat meal [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS 硝基呋喃类药物是一种光谱抗生素,对大多数革兰氏阳性菌和革兰氏阴性菌一及真菌等都有杀菌作用。硝基呋喃类代谢物广泛应用于畜禽和水产养殖,以治疗有大肠杆菌或沙门氏菌引起的肠炎、溃疡病等。硝基呋喃类药物在动物体内半衰期短,但是其代谢物可以和蛋白质紧密结合,残留时间长,甚至于经过高温、微波、烘烤等处理也无法降解[sup][/sup]。研究表明,硝基呋喃类及其代谢物对人体具有致癌、致畸胎副作用,欧盟已于1995年禁止在食用动物中添加硝基呋喃类药物[sup][/sup],中国卫生部在2010年就将硝基呋喃类药物硝基呋喃唑酮、呋喃它酮、呋喃妥因、呋喃西林列入违禁添加物质黑名单。对于硝基呋喃类药物的检测不能反映其在动物性体内真实的情况。目前对于硝基呋喃类药物的检测主要是对其代谢物的检测,主要方法有免疫分析法[sup][/sup]、液相色谱法[sup][/sup]、液相色谱-质谱法[sup][/sup]。液相色谱-质谱法灵敏度和选择性都比较高,对于基质复杂样品的检测具有明显的优势,是目前最常用的检测方法。 细胞的破碎与分离是代谢物的提取和分析中的重要步骤。高速动能处理技术是近一两年发展起来的一种新型的高科技的前处理方法。它通过一种极速的均质技术将鱼粉进行细胞破碎与分离,便于代谢物的提取。 鱼粉是由新鲜鱼类经过酶解或者蒸煮、胶磨、均质、喷雾干燥、过筛等一系列的程序生产的食品配料。其最大的特点是可以溶于水,既保留了天然鱼肉的风味,又富含营养。经过研究表明,鱼粉中粗蛋白的含量在50%以上,粗脂肪10%左右,氨基酸含量在40%-78%左右,其中人体必需氨基酸甘氨酸、精氨酸和脯氨酸含量较高。由于鱼粉在加工的过程中添加盐、防腐剂、增香剂等,导致样品基质复杂,在进行提取检测的过程中会出现分层不明显,油脂含量高等现象,难以保证检测过程中的准确性和灵敏性。目前关于鱼粉中硝基呋喃类代谢物的前处理几乎不涉及对样品的处理,因此建立一种新型的鱼粉高速动能处理技术对于硝基呋喃类代谢物的检测具有重要的研究价值和意义。本文通过高速动能前处理技术,同位素内标法,高温水解衍生快速处理。乙酸乙酯提取,正己烷除油,多重反应检测处理,从而建立一种样品前处理简单、提取速度快、净化效果好、选择性和灵敏度高的鱼粉中硝基呋喃代谢物的新型检测方法。[b]1实验部分[/b]1.1 主要仪器和试剂 Agilent1260-6460液相色谱-质谱联用仪(美国安捷伦公司);高速动能仪(美国);Sigma3K15离心机(德国);IKA RV10旋转蒸发仪(德国);恒温水浴锅;涡旋混合器XW-80A(中国);电子天平Secura224-1cn(瑞士);pH计;Milli-Q去离子水发生器(美国Millipore公司)。 硝基呋喃类药物的标准品;3-氨基-2-唑烷基酮(AOZ)、5-甲基吗啉-3-氨基-2-唑烷基酮(AMOZ)、1-氨基-2-内酰脲(AHD)、氨基脲(SEM)以及各自的同位素内标(纯度大于99%,均购至德国Dr.Ehrenstorfer GmbH);2-硝基苯甲醛;甲醇、乙腈;甲酸铵、甲酸(HPLC级,);乙酸乙酯、正己烷、四氯化碳、氢氧化钠、盐酸、磷酸氢二钾、磷酸二氢钾均为分析纯;超纯水(18.2ΜΩ)。1.2 标准溶液的配制 四种硝基呋喃代谢物标准贮备液:准确称取适量的硝基呋喃代谢物标准物质用乙腈配成1.0mg/ml的标准储配液,-18℃冷冻避光保存,有效期为三个月。四种硝基呋喃代谢物混合标准工作液(1.0μg/ml):取适量的硝基呋喃代谢物的标准贮备液用乙腈稀释成1.0μg/ml的混合工作使用液,-18℃冷藏避光保存,有效期为一个月,使用前回到室温左右。 氘代内标混合液储备液(0.1mg/mL):分别准确氘代内标物用乙腈溶解,配制成0.1mg/mL的标准储备液,-18℃冷冻避光保存,有效期为6个月。 氘代内标混合工作使用液(1mg/L):准确吸取一定量的氘代内标混合工作储备液用乙腈稀释成浓度为1mg/L的混合工作使用液,冷藏避光保存,有效期为1个月。 2-硝基苯甲醛衍生溶液:准确称取0.15g 2-硝基苯甲醛,用甲醇定容至10mL,现用现配。1.3 样品的前处理1.3.1 样品的水解和衍生 取一定的样品经过高速动能仪进行60s高速均质破碎。称取1.0g经过高速动能破碎的样品于50ml的离心管中,加入10ml的0.125mol/L的盐酸溶液,涡旋混匀1min,加入100ul2-硝基苯甲醛衍生试剂60℃恒温水浴衍生2h。1.3.2 提取 取出样品,冷却至室温。用2mol/L的氢氧化钠和1mol/L的磷酸氢二钾调节pH值至7.2左右。离心,收集上清液加入15ml的乙酸乙酯震荡30min,离心,转移上清液至另一离心管中,残渣继续用乙酸乙酯提取一次,合并清液。向上清液中加入15ml的正己烷震荡10min,离心,弃去正己烷层,重复上述操作一次。最后收集的清液于40℃条件下旋转蒸发至干,用1ml甲酸(0.2%)2.5ml的液态混合净化剂(正己烷:四氯化碳=1:1)转移至离心管中,超声,涡旋,离心,上清液过0.22μm滤膜过滤上机待测。1.3.3 UPLC色谱条件 Agilent超高效液相色谱柱:ZORBAX Eclipse Plus C[sub]18[/sub](1.8μm,2.1mm×50mm) 柱温:30℃;进样体积:2μL;流速:0.4ml/min;后运行:2min。梯度洗脱条件见下表1[align=center]表1 液相色谱洗脱条件[/align][align=center]Table 1 HPLCgradient elution program[/align][table][tr][td][align=center]时间(min)[/align][/td][td][align=center]A(0.1%甲酸水含乙酸铵)[/align][/td][td][align=center]B 乙腈[/align][/td][/tr][tr][td][align=center]0[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][tr][td][align=center]3.0[/align][/td][td][align=center]70[/align][/td][td][align=center]30[/align][/td][/tr][tr][td][align=center]5.1[/align][/td][td][align=center]0[/align][/td][td][align=center]100[/align][/td][/tr][tr][td][align=center]8.1[/align][/td][td][align=center]0[/align][/td][td][align=center]100[/align][/td][/tr][tr][td][align=center]8.2[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][/table]1.3.4 质谱条件 离子源:电喷雾离子源;监测方式:多重反应检测;毛细管电压:4000V;离子源温度:350℃;脱溶剂气压力:40psi;脱溶剂气流量:11 L/min。质谱条件见下表2。[align=center]表2 硝基呋喃代谢物MRM 分析参数[/align][align=center]Table 2 Analysis parameters of nitrofuran metabolites class MRM[/align][table][tr][td][align=center]project[/align][/td][td][align=center]parent ion[/align][/td][td][align=center]Daughter ion[/align][/td][td][align=center]energy/v[/align][/td][td][align=center]fragmentor/v[/align][align=center] [/align][/td][td][align=center]pattern[/align][/td][td][align=center]acceleration voltage/v[/align][/td][/tr][tr][td][align=center] AOZ[/align][/td][td][align=center]236.1[/align][/td][td][align=center]133.8[/align][/td][td][align=center]7[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AOZ[/align][/td][td][align=center]236.1[/align][/td][td][align=center]103.8[/align][/td][td][align=center]17[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]AOZ-D4[/align][/td][td][align=center]240[/align][/td][td][align=center]134[/align][/td][td][align=center]7[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AMOZ[/align][/td][td][align=center]335.1[/align][/td][td][align=center]291.2[/align][/td][td][align=center]6[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AMOZ[/align][/td][td][align=center]335.1[/align][/td][td][align=center]261.9[/align][/td][td][align=center]12[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]D5-AMOZ[/align][/td][td][align=center]340[/align][/td][td][align=center]296[/align][/td][td][align=center]8[/align][/td][td][align=center]110[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD[/align][/td][td][align=center]249[/align][/td][td][align=center]133.9[/align][/td][td][align=center]6[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD[/align][/td][td][align=center]249[/align][/td][td][align=center]104[/align][/td][td][align=center]18[/align][/td][td][align=center]100[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] AHD-13C3[/align][/td][td][align=center]252[/align][/td][td][align=center]134[/align][/td][td][align=center]7[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]SEM[/align][/td][td][align=center]209[/align][/td][td][align=center]192[/align][/td][td][align=center]6[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center] SEM[/align][/td][td][align=center]209[/align][/td][td][align=center]165.9[/align][/td][td][align=center]9[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][tr][td][align=center]SEM-C3,15N2[/align][/td][td][align=center]212[/align][/td][td][align=center]168[/align][/td][td][align=center]4[/align][/td][td][align=center]90[/align][/td][td][align=center]positive[/align][/td][td][align=center]3[/align][/td][/tr][/table][b]2结果与分析[/b] 2.1样品处理 鱼粉是一种基质复杂的粉状物质,大多数实验室是不经过样品处理的。硝基呋喃类在体内代谢迅速,代谢的部分化合物分子与细胞膜蛋白结合成结合态,也就是所谓的硝基呋喃代谢物。该代谢物比较稳定可以长期保持稳定状态,从而可以在人体内长期驻留。普通的处理很难将硝基呋喃类药物和蛋白质分离。本实验通过一种新型的高速动能仪是样品在极短的时间可以进行高速的均质使样品高速分散,造成组织和细胞破碎,代谢物的流出,有利于提取。对比经过高速动能处理后的样品的提取率和回收率,数据表明经过处理的样品很少出现乳化现象,提取率也高。本实验采用高速动能处理时间短、提取率高、很大程度避免了基质复杂提取过程中造成的乳化现象,非常适合于鱼粉中硝基呋喃代谢物类的检测。2.2 酸解、衍生化 四种硝基呋喃代谢物分子质量相对比较小,进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析时,在这个质量轴范围内易受低端分子离子的影响,同时这一段背景干扰比较大,对定性和定量分析造成一定程度的干扰。硝基呋喃代谢物在酸性条件下可以和蛋白质分离加入2-硝基苯甲醛衍生试剂后可以和其结合形成比较大的分子团避开小分子离子的干扰,获得较高的灵敏度和特异性。而衍生需要在一定的温度和时间条件下才可以,目前最常用的是37℃水浴振摇16h,耗时长,出现问题给检测工作造成很大的困扰。本实验对比不同的水浴温度和水浴时间,最终确定最佳条件,节省了检测时间 (见图 1)。[img=,690,349]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020909_01_2984502_3.png[/img]2.3线性和回收率 在对复杂基质进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析时常会出现基质效应,影响定量分析的准确性和精明度。本实验采用同位素内标法减少了外标法过程中存在的干扰,使定量变得更准确。分别对0.2μg/L、0.5μg/L、1.0μg/L、2.0μg/L、5μg/L的基质混合标准溶液进行测定,四种硝基呋喃代谢物的线性范围、线性方程、相关系数、检出限和定量限见下表3。结果表明:四种代谢物线性关系良好,检出限为0.3μg/L,相关系数均大于0.990。分别对低、中和高三个浓度进行加标回收,按照本方法进行提取、衍生和测定,平行测定3此,计算其回收率。采用统一添加水平的样品连续测定6次,计算其稳定性。结果见表4,四种硝基呋喃代谢物的平均回收率在91.7-105.2%之间,相对偏差小于3.10%,该方法适用性良好。[align=center]表3 鱼粉中四种目标物的线性范围、线性方程、相关系数、检出限和定量限[/align][align=center]Table Linear ranges,regression equations,LODs and LOQs for 4 analytes in samples[/align][table][tr][td]Analyte[/td][td]Linear ranges/(μg/L)[/td][td]regression equations[/td][td]R2[/td][td]LOD/(μg/Kg)[/td][td]LOQ/(μg/Kg)[/td][/tr][tr][td]AOZ[/td][td]0.2~5[/td][td]y=0.822736x-0.006286[/td][td]0.9997[/td][td]0.05[/td][td]0.20[/td][/tr][tr][td]AMOZ[/td][td]0.2~5[/td][td]y=2.515006x-0.022236[/td][td]0.9989[/td][td]0.05[/td][td]0.20[/td][/tr][tr][td]AHD[/td][td]0.2~5[/td][td]y=0.837220x+0.049223[/td][td]0.9997[/td][td]0.20[/td][td]0.50[/td][/tr][tr][td]SEM[/td][td]0.2~5[/td][td]y=2.210855x+0.033769[/td][td]0.9967[/td][td]0.20[/td][td]0.50[/td][/tr][/table][align=center]表4 样品的添加回收和精密度实验数据[/align][align=center]Table 4 Recoveries and repeatabilities for 4 analytes in sample[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Added/(μg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/(%,n=6)[/align][/td][/tr][tr][td][align=center]AOZ[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]91.7,94.3,97.2[/align][/td][td][align=center]2.34,2.56,2.92[/align][/td][/tr][tr][td][align=center]AMOZ[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]92.0,93.1,99.3[/align][/td][td][align=center]2.41,2.85,2.91[/align][/td][/tr][tr][td][align=center]AHD[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]94.6,98.7,105.2[/align][/td][td][align=center]2.69,2.19,3.10[/align][/td][/tr][tr][td][align=center]SEM[/align][/td][td][align=center]0.20,0.50,1.00[/align][/td][td][align=center]82.9,95.9,96.9[/align][/td][td][align=center]2.58,2.34,2.90[/align][/td][/tr][/table][b]3结论[/b] 本方法通过引用一种新型的处理样品的技术高速动能仪,使样品在极短的时间里达到高速分散,不仅可以提高提取率而且避免了后续提取过程中出现的乳化现象。本实验称取经过高速动能仪处理过的样品加入同位素内标,加酸使呋喃代谢物和蛋白质分开,加入衍生剂2-硝基苯甲醛在60℃水浴条件下震荡衍生2h,调pH值,正己烷除油,乙酸乙酯提取,液态净化剂净化,液相色谱-串联质谱分析,建立了检测鱼粉中四种硝基呋喃代谢物的高速动能处理方法。该方法采用一种新型的分散均质技术、优化水解衍生条件,具有耗时短、处理过程简单、提取率高、灵敏度好、检出限低等优点,能够满足国内外对硝基呋喃代谢物限量的要求。
最近做复硝基酚钠的检测,做液质方法开发质谱条件做好了,可是进液相竟然邻硝基苯酚钠不出峰,不知道怎么回事?按说这两个同分异构体,应该性质差不多吧离子对都完全相同怎么会一个出峰,一个不出峰呢?看文献中也提到检出限的问题,邻硝基苯酚钠检出限更高一些可能是这个物质不容易电离的缘故吧不知道有什么方法可以促进其电离》
HJ 1150-2020 《水质 硝基酚类化合物的测定》标准中 [font=Times New Roman][color=#000000]2,4-[/color][/font][font=宋体][color=#000000]二硝基酚、2,6-二硝基酚的响应值特别低,在该物质出峰时间处加电压效果也不是很理想。请教各位做过本标准的版友,有什么好的方法可以提高二者的响应吗?[/color][/font]
不知上海有哪家试剂公司有2,6-二硝基酚卖,急需!!!
近日,环保部就《水质硝基酚类的测定 气相色谱-质谱法》(征求意见稿)、《水质 二氧化氯的测定 碘量法》(征求意见稿)两项国家环境保护标准发布征求意见的函。 其中,《水质硝基酚类的测定 气相色谱-质谱法》为首次发布,《水质 二氧化氯的测定 碘量法》则是对《水质二氧化氯的测定碘量法(暂行)》(HJ 551-2009)的修订。 硝基酚类是危害环境的有机污染物,可在水生生物和人体中残留和浓缩,具有高毒性和致癌性,4-硝基酚被我国列入环境优先监测污染物监测名单中,但目前我国尚没有关于水质硝基酚测定方面的标准分析方法。 气相色谱质谱法在有机污染物分析方面具有分辨率高、定性准确等优点,因此,该标准采用了液液萃取、固相萃取气相色谱质谱法测定水中硝基酚类方法,经验证,可以满足水质中硝基酚类化合物测定特性指标的要求。
请问如何对样品检验确认是否含有六硝基菧(HNS)和二硝基重氮酚(DDNP)?如显色反应或沉淀反应。
[align=right][b]SGLC-GC/MS-020[/b][/align] [b]摘要:[/b]本文建立了12种硝基酚类化合物测定的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]方法。参照HJ 1150-2020中色谱方法,采用色谱柱 SH-I-5SilMS对12种硝基酚类化合物进行分析,岛津[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-TQ8050NX[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪进行检测。结果表明,12种硝基酚类化合物峰形对称,重现性好,满足标准要求。本方法可为12种硝基酚类化合物的测定提供参考。 [b]关键词:[/b]水质 硝基酚类 SH-I-5SilMS [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url] [b]1. 实验部分 1.1 实验仪器及耗材[/b] 岛津[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-TQ8050NX[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪; 色谱柱:SH-I-5SilMS(30 m×0.25 mm×0.25μm;P/N:221-75954-30); 纯水机:PR-FP-0120α-MT1(+ 60L水箱 + 取水器); SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05); [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01); SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02); SHIMSEN Pipet PMII-100(P/N:380-00751-04); SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。 [b]1.2 混合标准使用液的制备[/b] 取市售硝基酚类混合标准品适量,用二氯甲烷稀释定容至2.0mg/L,作为混合标准使用液。 [b]1.3 分析条件 GC条件[/b] 毛细管柱:SH-I-5Sil MS(30 m×0.25 mm×0.25μm;P/N:221-75954-30) 程序升温:初始温度50℃,保持5min,以8℃/min升温到250℃,保持4min 载气:He 载气控制模式:恒线速度 流速:1.0ml/min 进样口温度:220 ℃ 进样量:1μL 进样方式:不分流进样 [b]质谱条件[/b] 电离模式:电子轰击电离(EI) 电子轰击能量:70 eV 离子源温度:230 ℃ 接口温度:260 ℃ 溶剂延迟:3min 数据采集模式:SIM 各化合物SIM参数见下表 [img=SHIMSEN Styra HLB]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-020_01.png[/img][font=arial, &][size=12px][/size][/font] [b]2. 实验结果[/b] 按照上述色谱条件(1.3)进行采集,混合标准使用液色谱图如下: [b]混合标准使用液[/b] [img=SHIMSEN Styra HLB]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-020_02.png[/img][font=arial, &][size=12px][/size][/font] [b]重现性数据[/b] [img=SHIMSEN Styra HLB]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-020_03.png[/img] [b]3. 结论[/b] 本文建立了12种硝基酚类化合物测定的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]方法。参照HJ 1150-2020的方法,采用色谱柱SH-I-5SilMS对12种硝基酚类化合物进行分析,岛津[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-TQ8050NX[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪进行检测。结果表明,12种硝基酚类化合物峰形对称,重现性好,满足标准要求。本方法可为12种硝基酚类化合物的测定提供参考。
请问大家用HPLC测对硝基苯酚的标样时,有没有出来两个峰? 看文献有的是说在溶剂峰出来前的那个峰是脂肪族化合物的峰,后面那个峰是对硝基酚的。按理论来说,标样应该只有一个峰的。
事件回放:“丈夫没有了,两个孩子也还在医院里,这两天我都不知道怎么过来的。”刘从兰用手擦去眼角的眼泪,静静地望着刚做完血透的小女儿琳琳(化名)。她说,现在一点都不敢想象今后的生活该怎么过下去,怕自己会承受不了。 9月13日,东阳市画水镇发生一起因废塑中毒事件,目前已造成3人死亡、17人住院治疗的严重后果。[font=黑体][size=4]何谓二硝基苯酚?[/size][/font][color=#00008B]分子式2,4-(NO2)2C6H3F。 2,4-二硝基氟苯为淡黄色晶体; 熔点25.8℃,沸点 296℃,密度1.4718克/厘米3(84℃); 溶于乙醇、苯、丙二醇等。 2,4-二硝基氟苯主要由2,4-二硝基氯苯与氟化钾在硝基苯中反应制得 2,4-二硝基氟苯是一种重要的分析试剂,用来鉴定有机化合物中的氨基,尤其是用于蛋白质或多肽的N-端残基分析。鉴定时,2,4-二硝基氟苯与肽链的游离氨基作用,生成2,4-二硝基衍生物。将其水解后,末端氨基酸的N-(2,4-二硝基苯基)衍生物常为亮黄色结晶,易与其他氨基酸分离。该方法结合其他方法,可确定蛋白质或多肽氨基端碳链的结构。由F.桑格于1945年提出,故称桑格法 此外,它在碳酸氢钠溶液中与醛糖的肟反应时可发生降解,生成次级醛糖、2,4-二硝基苯酚和氢氰酸,故可用于醛糖的分析。2,4-二硝基氟苯能使皮肤糜烂,使用时应注意。[/color]&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&77[color=#DC143C]1)作为一名专业的分析工作者,如果从自己的专业角度去分析。我们使用什么方法和仪器可以检测出二硝基苯酚?2)遇到这样的事情如何加强自我预防?3)在我们平时的分析工作中,我们会遇到那些有毒的化学试剂或者样品,我们如何做好自我防备。欢迎大家讨论。。。。[/color]
维权声明:本文为ncicjxb原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。摘要 通过正交实验和验证实验确定极谱测定苯胺中微量硝基苯的最佳条件,同时对悬汞电极、静态滴汞电极、滴汞电极三种电极模式以及微分脉冲极谱和经典直流极谱两种极谱类型进行了实验比较,得出相应结论。关键词 极谱 硝基苯 苯胺1. 通过正交实验和验证实验确定了极谱测定苯胺中硝基苯的最佳条件,即除氧时间180S、冰醋酸2 mL、10%乙醇10mL。在实验中发现随着冰醋酸加入量的增加,硝基苯的半波电位正移,2 mL时为-0.4V,4 mL时为-0.35V,6 mL时为-0.33V。2. HMDE、SMDE、DME三种电极模式下的硝基苯峰电流与含量线性关系显著,HMDE线性范围在0.2-600 mg/L,SMDE、DME线性范围在0.1-600 mg/L。灵敏度从大到小排序为DME、SMDE、HMDE,汞耗从大到小顺序也为DME、SMDE、HMDE。在最低检测量上HMDE约在0.2 mg/L,而SMDE、DME约在0.1 mg/L。所以在选择电极模式时要综合考虑测定灵敏度、汞耗和最低检测量几方面。3. 虽然微分脉冲极谱和经典直流极谱的硝基苯峰电流与含量线性关系都显著,线性范围都在0.2-600 mg/L,但同样条件下微分脉冲极谱的电流大约是经典直流极谱的4倍,即微分脉冲极谱的灵敏度约是经典直流极谱的4倍,所以微分脉冲极谱应当被优先选择。
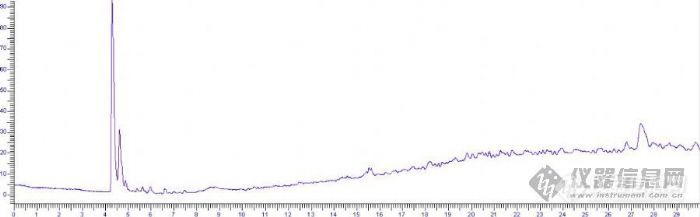
Pe500 进样口:250度,弱极性柱:110度(6分),5度每分到180度(10分)ECD350度分析间硝基氯苯,甲醇做溶剂,用的标准品。图1是甲醇洗针后针的本底图。图2是吸间硝基氯苯溶液上气体后,推针2下,再吸空气的色谱图,应该只有间硝基氯苯的峰啊,怎么在14和29分钟出现两个峰?高手解答! [img]http://ng1.17img.cn/bbsfiles/images/2008/09/200809031646_106955_1775690_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2008/09/200809031649_106956_1775690_3.jpg[/img]
各位高手帮帮忙,我们实验室现在用对硝基苯酚做指示剂,但是很不稳定,在空气中置放一会儿就变色了,想问下究竟是什么原因使它变色的?有什么方法可以延缓呢?谢谢各位了
称取粉碎后的样品2g(精确到0.05g)待测样品于50mL离心管中,加入5mL0.2mol/L盐酸溶液,和0.15mL2-硝基苯甲醛溶液,旋涡振荡~1min。避光置于37℃恒温水浴振荡器中过夜(约16hr)。7.3 样品净化待恢复至室温,加入5mL 4mol/L 磷酸氢二钠溶液,调节pH直至7.0~7.5,加入4mL乙酸乙酯,漩涡振荡~1min,4000r/min离心5~10min,取上层清液转移至10mL玻璃离心管管中;再加入4mL乙酸乙酯,漩涡振荡~1min,4000r/min离心5~10min,取上层清液转移至同一10mL玻璃离心管管中。在氮气氛围下,40℃吹干溶液。精确加入1.0mL甲醇水溶液(甲醇:水=5:95)溶解残留物,过0.45um滤膜,过滤后上机。拿这个前处理的方法做面包糠,预裹粉等辅料,做到加乙酸乙酯离心后,会形成果冻状,乙酸乙酯层吸不出来?是什么原因呢?怎么解决?
请问专家循环水中如何测定苯酚和硝基酚类物质的含量?
如何用方波溶出伏安法测硝基酚????
如何用方波溶出伏安法测硝基酚??
最近两天多宝鱼中硝基呋喃的事情比较热,有几个大城市多宝鱼都下架了,不过我觉得其他鱼或水产类动物肯定还存在类似问题,不知道大家有没有什么新发现
如题,有做过废水中硝基苯酚测定的版友么,能不能分享一下你的方法,做的效果如何。先谢谢了~
最近看了一篇文献Graphene oxide and reduced graphene oxide as novel stationary phases via electrostatic assembly for open-tubular capillary electrochromatography(Electrophoresis2013,34,1869–18760)文中以氧化石墨烯为电色谱固定相来分离邻、间、对硝基苯酚,其pKa分别为8.39, 7.15, 和7.22 文中选择pH7.0的磷酸缓冲液为流动相,说是可以得到基线分离。但是,pH7.0下间硝基苯酚(7.15)和对硝基苯酚(7.22)都是以负离子和中性分子两种形式存在,邻硝基苯酚以分子形式存在,最后得到的邻硝基苯酚的峰中会不会掺杂有间硝基苯酚和对硝基苯酚的分子形式??? 这和氧化石墨烯有关吗? 氧化石墨烯在试验中的作用?对硝基苯酚的三种位置异构体的作用大小不一样吗? 谢谢各位
如题,雨木霖专家来领分!
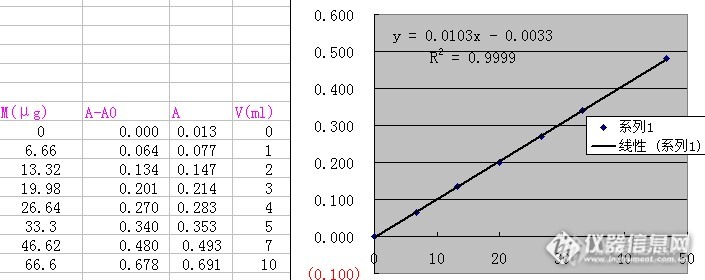
硝基苯类化合物主要存在于染料、炸药、制革等的工业废水中。排入废水中对人体危害极大,产生毒性作用,引起神经系统的症状,贫血以及肝脏疾病。参照《水和废水监测分析方法》进行硝基苯类中一硝基和二硝基化合物的测定。【方法原理】——在含硫酸铜的酸性溶液中,由锌粉反应产生的初生态氢将硝基苯还原成苯胺,经重氮偶合反应生成紫红色染料,在545nm波长进行比色测定。【方法适用范围】——适用于测定染料、制药、皮革及印染等行业废水中的硝基苯类化合物,最低检出浓度为0.2mg/L。【方法具体操作】1、先绘制标准曲线:1.1 吸取1.00ml硝基苯使用溶液于50ml锥形瓶中,加水至20ml,加入浓盐酸2.0ml,锌粉0.5g,10%CuSO4溶液两滴,摇匀。放置15min,用慢速滤纸过滤,滤液收集于50ml容量瓶,稀释至标线,混匀。1.2 吸取0.0、1.0、2.0、3.0、4.0、5.0、10.0ml分别置于25ml比色管中,加水至10ml刻度线,+10%NaOH溶液至出现白色絮凝状沉淀(pH 5),加水至标线,摇匀。+1ml20%硫酸氢钾溶液(调节pH),待白色沉淀消失,+5% NaNO2溶液1滴,摇匀,放置3min。+2.5%氨基磺酸氨溶液0.5ml,以除去剩余的NaNO2,充分摇匀,放置3min。待气泡除尽,+2%盐酸奈乙二胺溶液(N-(1-萘基)乙二胺)1.0ml,加塞摇匀。1.3 放置30min,用10mm比色皿,于545nm波长处,以水为参比,测量其吸光度,绘制标准曲线即可。2、再是关于实际样品的测定2.1 样品测定 ① 取适量水样于锥形瓶中,加水至20ml,下面步骤同1.1。 ② 取与上述相等量的水样于50ml容量瓶中,+浓HCL 2.0ml,10% CuSO4 2滴,加水至标线,混匀。 ③ 分取上述①、 ②溶液各10ml(不得超过10ml),置于25ml比色管中,与绘制校准曲线步骤相同,水为参比,测量A。 由①、②所测得的吸光度减去空白吸光度后的差值,分别为水样中硝基苯类和苯胺类的吸光度值。2.2 空白试验 即取20ml实验用水于锥形瓶中,步骤及其他试剂用量与样品测定相同,测量空白吸光度。【注意事项】1、最适宜的显色温度在22~30℃,当低于此温度时,尤应注意校准曲线跟水样同时进行操作。2、加10%氢氧化钠溶液于经还原操作的水样中,当pH调至4~5时,溶液可能出现絮凝状沉淀,而不经还原操作的水样无絮凝沉淀。3、水样经还原操作过滤时,应使用慢速滤纸。以上都是方法上的。下面是本人自己摸索后的感悟及成果,如有错误请指出:关于温度,不管是测苯胺还是硝基苯,最好的温度就是30度,曲线线性很好,都达到0.9999.加氢氧化钠絮凝的话,不能加过多,有白色出现就好,不然后面还得溶解就很难;给本人的感觉,这个步骤只是验证有没有被还原而已的。水样还原后要进行过滤,采用慢性滤纸应该是为了多点时间还原的;我就把还原的时间拉长,用中速的过滤的。做过对比,感觉没什么影响。下面是我做的两条曲线:http://ng1.17img.cn/bbsfiles/images/2013/08/201308061739_456454_2721409_3.png2013.05.08http://ng1.17img.cn/bbsfiles/images/2013/08/201308061739_456455_2721409_3.png
[color=#444444]真心请教各位色谱高手,有哪位知道[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测邻硝基苯酚的色谱条件是什么啊?我按照查的文献和实验书上的条件做可是都没有峰,我都不知道怎么样开始摸索改变条件,主要条件是柱温、进样温度、检测器温度、流速吗?那各条件设定的依据是什么啊?非常谢谢~~[/color]
[color=#444444]真心请教各位色谱高手,有哪位知道[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测邻硝基苯酚的色谱条件是什么啊?我按照查的文献和实验书上的条件做可是都没有峰,我都不知道怎么样开始摸索改变条件,主要条件是柱温、进样温度、检测器温度、流速吗?那各条件设定的依据是什么啊?非常谢谢[/color]
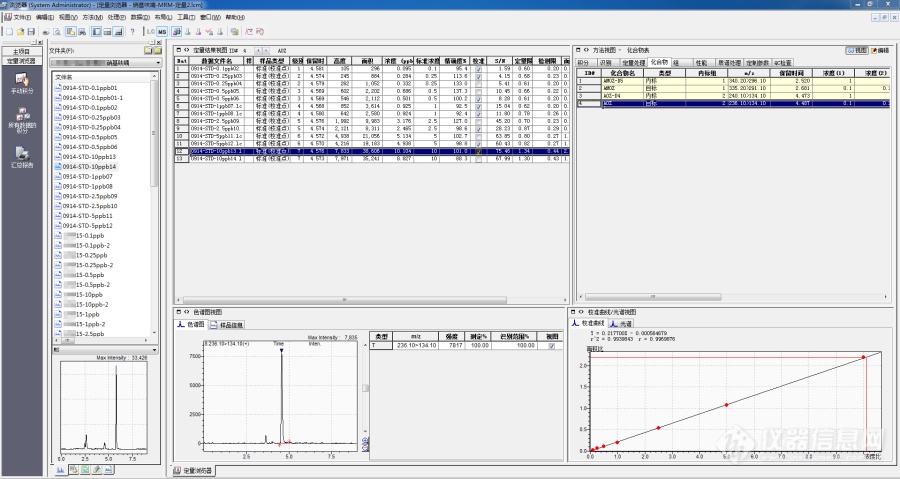
[b][b]液相色谱-串联质谱法测定草鱼中硝基呋喃类代谢物AOZ残留量[/b]摘要[/b]: 本文根据农业部783号公告-1-2006水产品中硝基呋喃类代谢物残留量的测定 液相色谱-串联质谱法来检测草鱼中硝基呋喃类代谢物AOZ的残留。样品经水解、衍生化和净化后,采用多反应监测模式测定。通过化合物的保留时间、定性离子和定量离子的筛查与确证,实现对草鱼中硝基呋喃类代谢物AOZ的定性与定量。结果表明,其线性系数大于0.99,在2.5mg/kg和4.5mg/kg添加水平下,AOZ回收率在95%-100%之间,相对标准偏差(RSD)小于15%。样品经水解、衍生化和净化后,以流动相A:0.002mol/L的醋酸铵溶液,B:甲醇进行梯度洗脱,C18(100mm×2.1mm,i.d,5um)色谱柱分离,采用正离子多反应监测( MRM) 模式检测,基质匹配标准溶液定量。线性范围内相关系数均大于[color=#ff0000] [/color]0. 99。加标回收率在95.00%-100% 之间,相对标准偏差在6.19%- 8.43% 之间,结果表明,该方法简便、快速、灵敏度高、重现性好,可测定草鱼中硝基呋喃类代谢物AOZ的残留。[b]关键词[/b]: 高效液相色谱 - 串联质谱 硝基呋喃代谢物AOZ 草鱼;[b]Abstract[/b]:In this paper, AOZ residue of nitrofuran metabolites in grass carp was detected by liquid chromatography-tandem mass spectrometry according to the determination of nitrofuran metabolites residues in aquatic products in the ministry of agriculture no.783 bulletin 1-2006. After hydrolysis, derivatization and purification, the samples were determined by multi-reaction monitoring. AOZ, a nitrofuran metabolite in grass carp, was qualitatively and quantitatively determined by retention time, qualitative and quantitative ion screening. The results showed that the linear coefficient was greater than 0.99, the recovery of AOZ was between 95% and 100% and the relative standard deviation (RSD) was less than 15% at the addition levels of 2.5mg/kg and 4.5mg/kg.After the samples were hydrolyzed, derivated and purified, the samples were separated by gradient elution in mobile phase A:0.002mol/L ammonium acetate solution,B: methanol, and separated by C18(100mm×2.1mm, i.d,5um) chromatographic column. Positive ion multireaction monitoring (MRM) mode was adopted for detection, and the matrix matched standard solution quantification. The correlation coefficients in the linear range are all greater than 0.99. The standard recovery was between 95.00% and 100%, and the relative standard deviation was between 6.19% and 8.43%.[b]Key words[/b]:ultra performance liquid chromatography-tandem mass spectrometry;nitrofuran metabolites-AOZ Grass carp.[b]1 实验部分[/b]1.1 仪器与试剂液相色谱—质谱联用仪( 岛津[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]-8030) 超纯水系统(WP-UP-YJ-20, 沃特浦) 分析天平(Quintix 2102-1CN,赛多利斯公司);旋涡混合器(VORTEX 3,IKA公司);恒温水浴振荡器(江苏安普SHA-C);离心机(湖南湘仪H-2050R);甲醇(色谱纯,默克股份两合公司) 醋酸铵( 分析纯,天津市致远化学试剂有限公司);2-硝基苯甲醛(色谱纯,天津市百世化工有限公司);二甲基亚砜(分析纯,天津市百世化工有限公司);磷酸氢二钾(分析纯,天津市百世化工有限公司);乙酸乙酯(分析纯,天津市泰兴试剂厂);农药标准物质: 均来自农业部环境保护科研监测所(天津);实验用水为经沃特浦超纯水系统处理后的超纯水。1.2 [b]样品处理[/b]1.2.1[b] 样品水解与衍生化[/b] 准确称取样品2.0g(精确至 0.01 g) 到50 mL 离心管中,加入0.05mL的100ng/mL混合内标(AOZ-D[sub]4[/sub]),旋涡混合50s,再加入5mL盐酸溶液(0.2mol/L)和0.15mL的2-硝基苯甲醛溶液(0.05mol/L),涡旋振荡50s后,置于恒温水浴振荡器中37℃避光震荡16h。1.2.2 [b]样品的提取净化[/b]取出离心管冷却至室温,加入3-5mL磷酸氢二钾溶液(1.0mol/L),调节pH至7.0-7.5,加入4mL乙酸乙酯,涡旋振荡50s,4000r/min离心5min,取上层清液转移至10mL玻璃离心管中;再加入4mL乙酸乙酯重复上述操作,合并上清液于40℃下氮气吹干。加入1.0mL甲醇溶液(甲醇:水=5:95(V:V))涡旋振荡溶解残留物,过0.45um滤膜,待测。1.2.3 [b]溶液配制 [/b](1)[b]标准储备液。[/b] 分别取上述标准品(100.0 ug/mL 溶液),用甲醇稀释成 10 ng/mL 和100 ng/mL的标准储备液。(2)[b]标准工作溶液。[/b] 分别吸取上述标准品10ng/mL的标准储备液0.010mL、0.025mL、0.050mL、0.10mL和100ng/mL的标准储备液0.025mL、0.05mL、0.10mL于7个50mL离心管中,不加样品,按照1.2.1和1.2.2步骤操作,按照1.3测定。1.3 [b]色谱 - 质谱条件[/b]1.3.1 [b]色谱条件 [/b] 色谱柱:C18柱,(100mm × 2.1mm,5μm ) 柱温为40℃;进样量为20μL。流动相:A.0.002mol/L的醋酸铵溶液,B:甲醇 色谱洗脱条件见表 1。[align=center]表 1 高效液相色谱梯度洗脱条件[/align][table][tr][td][align=center]时间,min[/align][/td][td][align=center]A,%[/align][/td][td][align=center]B,%[/align][/td][td][align=center]流速,mL. [/align][/td][/tr][tr][td][align=center]3.5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5[/align][/td][td][align=center]15[/align][/td][td][align=center]85[/align][/td][td][align=center]0.25[/align][/td][/tr][tr][td][align=center]5.1[/align][/td][td][align=center]76[/align][/td][td][align=center]24[/align][/td][td][align=center]0.25[/align][/td][/tr][/table]1.3.2[b]质谱条件[/b]离子源: 电喷雾离子源ESI 扫描模式为正离子扫描 雾化气流量3L/min;干燥气流量15L/min;加热块温度250℃;DL温度250℃;CID气230kPa;IG真空度1.9×10[sup]-3[/sup]pa;PG真空度7.5×10[sup]1[/sup]pa;检测方式为多重反应监测。其他质谱参数见表2。 [align=center]表 2 反应监测的质谱采集参数[/align][table][tr][td][align=center]化合物[/align][/td][td][align=center]母离子m/z[/align][/td][td][align=center]子离子m/z[/align][/td][td][align=center]碰撞能量(v)[/align][/td][/tr][tr][td=1,2][align=center]AOZ[/align][/td][td][align=center]236[/align][/td][td][align=center]104[/align][/td][td][align=center]19[/align][/td][/tr][tr][td][align=center]236[/align][/td][td][align=center]134*[/align][/td][td][align=center]22[/align][/td][/tr][tr][td][align=center]AOZ-[/align][/td][td][align=center]240[/align][/td][td][align=center]134*[/align][/td][td][align=center]14[/align][/td][/tr][tr][td=4,1]注:*为定量碎片离子[/td][/tr][/table][b]1.4 添加回收实验[/b] 准确称取不含上述药物的草鱼样品2.0g,分别以2.5mg/kg和4.5mg/kg2个水平进行添加回收实验,重复5次,按照上述实验方法测定,计算添加回收率和相对标准偏差。[b]2 结果与讨论2.1线性回归方程[/b]AOZ的基质标准曲线为 y = 0.217700 x -0.000564679( r = 0.9969876,r[sup]2[/sup]=0.9939843) 如图3 [align=center]图3(标准曲线AOZ)[/align][img=图片3 标准曲线,690,367]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100829478357_6345_3416090_3.png!w690x367.jpg[/img][b]2.2方法的准确度、精密度[/b]在空白草鱼中分别进行 2.5和4.5 mg / kg 2 个水平的加标回收试验,每个水平重复测定 5 次,计算加标回收率和相对标准偏差。由表 5 可知,在 2.5和4.5mg / kg 2个添加水平下,AOZ的平均回收率分别为 99.648% 和95.63% ,相对标准偏差分别为 8.43% 和6.19%,方法显示出良好的准确度和精密度,可以满足试剂样品中农药残留的检测要求。[align=center]表 5 AOZ在草鱼中的回收率和相对标准偏差[/align][table][tr][td][align=center]农药[/align][/td][td][align=center]添加浓度( mg / kg)[/align][/td][td][align=center]平均回收率( %)[/align][/td][td][align=center]重复 1[/align][/td][td][align=center]重复 2[/align][/td][td][align=center]重复 3[/align][/td][td][align=center]重复 4[/align][/td][td][align=center]重复 5[/align][/td][td][align=center]平均值相对标准偏差( % )[/align][/td][td][align=center]标准曲线[/align][/td][td][align=center]相关系数r2[/align][/td][/tr][tr][td]AOZ[/td][td]2.5[/td][td]99.648[/td][td]2.517[/td][td]2.261[/td][td]2.663[/td][td]2.723[/td][td]2.292[/td][td]8.43[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][tr][td]AOZ[/td][td]4.5[/td][td]95.63[/td][td]4.223[/td][td]4.768[/td][td]4.043[/td][td]3.870[/td][td]4.613[/td][td]6.19[/td][td][align=center]Y=0.217700X-0.000564679[/align][/td][td]0.9939843[/td][/tr][/table][b]2.3实际样品分析[/b] 用本方法对市场销售的2份草鱼进行硝基呋喃类代谢物AOZ检测,均未检出,见图6。 [align=center]图6[/align][img=图片6,690,366]https://ng1.17img.cn/bbsfiles/images/2019/10/201910100830145934_7979_3416090_3.png!w690x366.jpg[/img][b]3、结论[/b]本研究通过对样品前处理方法、质谱条件和色谱条件的优化,建立了液相色谱-串联质谱法测定草鱼中AOZ残留量的分析方法,该方法前处理简单、快速、回收率高,方法的灵敏度、准确度和精密度等均满足农药残留分析的要求,适用于大量样品的快速检测。[b]参考文献[/b]高洁,朱莉萍等.超高效液相色谱-串联质谱法测定多脂肪类动物源性食品中硝基呋喃代谢物. 食品安全质量检测学报2018年第9卷第6期,2018.[b] [/b]
求助各位高手!对硝基苯酚在水溶液中不稳定,容易电离成醌式结构而使水溶液颜色变深,如何保持它的水溶液长期稳定不变色,而且当水溶液的氢离子浓度改变时,这种平衡发生移动,可以有哪些方法保持呢?谢谢各位帮忙







 我要推广仪器
我要推广仪器
 下载APP
下载APP