请问有人做APEOs或者邻苯二甲酸酯类的吗?做什么样品,样品怎么处理?检测条件是什么?
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质检测氨基甲酸酯类农药:按照HJ827-2017用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质做氨基甲酸酯类农药,可是用水配制标准恶虫威不出峰,但是相同的条件用甲醇配制的标液恶虫威能正常出峰,恶虫威在水中的可溶的,请问是什么原因造成的?
[em09511]果蔬汁中对羟基苯甲酸乙酯能力验证,我们用液相做的,粗测大概在157和252mg/kg,不知有没有大家可不可以交流下啊?!
求对羟基苯甲酸甲/乙/丙/丁酯 USP OR EP方法我没找到,不知道是否英文输入错误,还是没有相关标准,谢谢
有参加德国DRRR组织的护理产品中对羟基苯甲酸甲酯,对羟基苯甲酸乙酯,对羟基苯甲酸丙酯,对羟基苯甲酸丁酯,苯氧基乙醇,苯甲酸,山梨酸,甲醛,甲基异噻啉酮 能力验证的吗。可以加Q群39177650交流,或在这留言哦。测试轮编号2010206care Products-methylparaben, ethylparaben, propylparaben, butylparaben, phenoxyethanol, benzoic acid, sorbic acid , formaldehyde,methylisothiazolinone
求助 邻苯二甲酸二乙酯在液谱上做内标物的应用实例谢谢!!!
[em09504]测定磷酸三丁酯,以邻苯二甲酸二丁酯做内标物,以二甲苯作溶剂,峰会重叠吗?http://www.instrument.com.cn/bbs/shtml/20030612/218050/以二甲苯作溶剂,大家觉得按上面的帖子可行吗?邻苯二甲酸二丁酯使用分析纯的可以吗?还是要用色谱纯的?
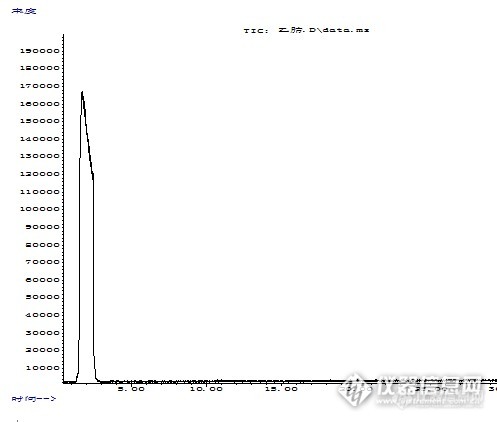
我是准备测定白酒中甲酸乙酯的含量。按着试验方法先试着做一次标品。试验方法是按《SN/T 0285-2012出口酒类中氨基甲酸乙酯残留量检验方法》 我想请问一下前辈们见过这样的情况么,是什么原因可能导致的这种情况,有什么解决的办法么?一、溶剂走样测定条件(1)色谱参考条件色谱柱:HP—WAX 石英毛细管柱: 30m×0.25mm(内径)×0.25 μm(膜厚),或相当色谱柱(实际所用柱子为柱子DB-1701ms)。柱温:初温 60℃,保持 1min, 以5℃/min升至 150℃保持2min。以20℃/min升至240℃保持10min。载气:氦气,纯度≥99.999%,流速 1 mL/min。进样口温度:220℃;进样量:1μL;进样方式:无分流进样,1min开阀。溶剂延迟:4min(2)质谱参考条件电离方式:EI源, 70eV。离子源温度:230℃。氨基甲酸乙酯选择监测离子(m/z):44、62、89,定量离子62。D5-氨基甲酸乙酯选择监测离子(m/z)44、64、76,定量离子64。测定后出图如下:http://ng1.17img.cn/bbsfiles/images/2014/04/201404171323_496455_2863381_3.jpg二、1μg/ml氨基甲酸乙酯走样测定条件(1)色谱条件同上。溶剂延迟:14min(14min时没有峰出现,后来改为4min)[font
大家好,最近在做PAEs的检测,参照标准是GB/T 21928-2008 食品塑料包装材料中邻苯二甲酸酯的测定,买的是16种PAEs的混标,仪器是岛津GC-MS。现在做标样时0.8个ppm出的峰很少,而且强度很低。大家做PAEs时标样能做到的级别是多少啊?用的是哪家仪器?(21911的检测方法同这个方法基本差不多)
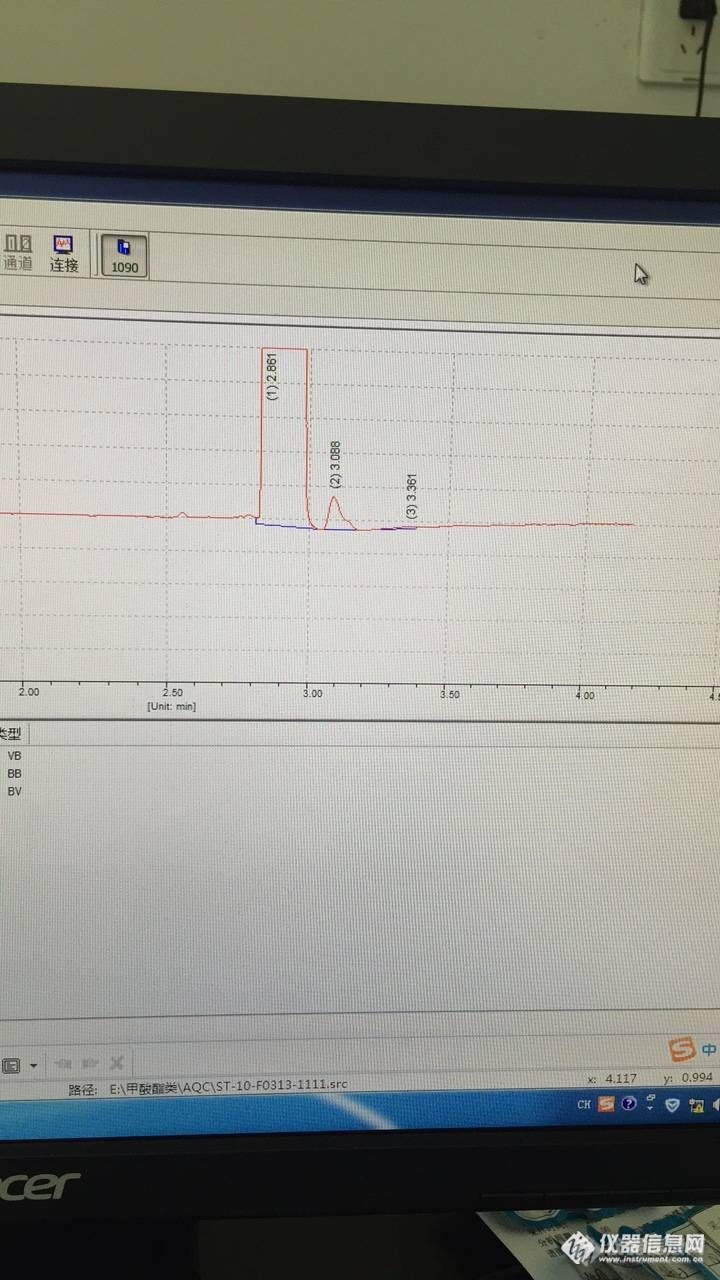
我在前一天做的乙酸甲酯、乙酸丙酯、乙酸戊酯和乙酸异丁酯的混标,都没问题,曲线做的也很好,之前摸条件的时候看到甲酸甲酯和甲酸乙酯会和乙酸酯类有重叠,所以乙酸酯类和甲酸酯类就打算分开来做,之前甲酸酯类摸条件的时候峰都是能出来的,只不过摸条件的时候浓度比较大,估计有2000mg/L。在第一天做完乙酸酯类混标曲线之后第二天做甲酸酯类,就发现甲酸甲酯的峰出不来,从曲线最低点的没有峰到曲线最高点的峰面积只有200,我接着重新开了一只纯品重新称重重新配曲线,还是一样,峰就是出不来,后来做了甲酸甲酯单项目的曲线,也是出不来峰,我一开始做乙酸酯类的条件是进样口220°,柱箱90°,检测器250°,走的恒温,(小白,不会程序升温 - -|||)后来做的不好,我柱箱温度从65°到90°都试过了,都不行,分流比一直是30。http://ng1.17img.cn/bbsfiles/images/2017/03/201703131502_01_3079836_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703131503_01_3079836_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703131503_01_3079836_3.jpg
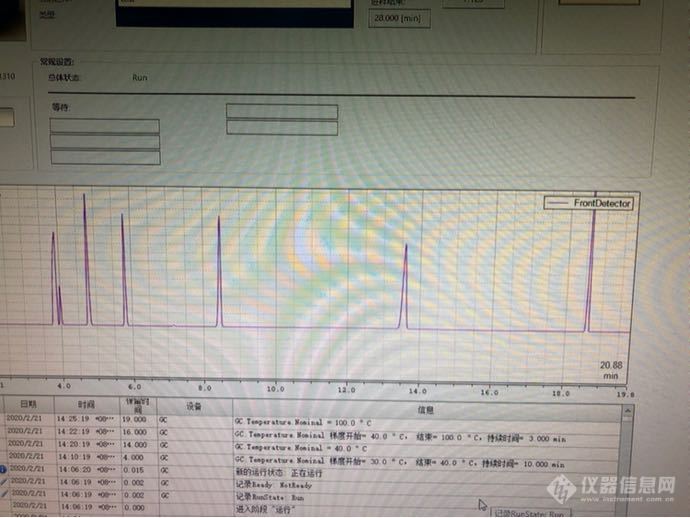
我买的坛墨的7种饱和脂肪酸酯类化合物混标!甲酸甲酯 乙酯 乙酸甲乙丙丁戊酯。现在问题来了。我甲酸乙酯和乙酸甲酯一直分不开 。下面是我条件:30 度保持4min 以1 度/min 升到40度 30度/min到100。后面都没问题 甲酸甲酯我温度低一点也能和cs2分开。可是甲酸乙酯和乙酸甲酯...分不开。那我怎么做呢?是不是要单独做一个乙酸甲酯曲线方法验证。柱子是 ffap 30 0.32 0.25[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/02/202002211425479329_4319_2990176_3.png[/img]
邻苯二甲酸酯类检测,进标准浓度是多少?保留时间有没有飘移?浓度越大,保留时间越短,怎么设置sim时间,做不同的标准,尤其是1ppm、5ppm、10ppm、50ppm、100ppm标准曲线
我是刚刚接触气质联用仪的新手,准备测定白酒中甲酸乙酯的含量。 今天按着试验方法先试着做一次标品(浓度是1ug/mL),我在选用溶剂延迟14min时,总离子图中没有峰出现,试验方法又说出峰时间在18min左右,然后我选用了溶剂延迟4min后,在第5-6min左右就出现了峰,质谱图里面有EC,但是还有溶剂乙腈,我想请教一下您是什么原因。 请大家帮帮我分析一下是什么问题!谢谢了! 我用的是安捷伦的气质仪,试验方法是按《SN/T 0285-2012出口酒类中氨基甲酸乙酯残留量检验方法》
有用GC/MS做白酒中的氨基甲酸乙酯的朋友吗?
最近准备做氨基甲酸乙酯,用之前的标准品,用之前的方法做不出峰来,用1ppm的全扫也不出峰之前是同事做的,不知道不出峰会是什么原因?是标准品不稳定分解掉了,还是现在仪器状态不如以前了?有没有做过氨基甲酸乙酯的朋友,请多多指教!谢谢
做邻苯二甲酸酯时,DEEP中,进5个标准,第一个标准72离子浓度最大,到第二个标准时,221离子浓度最大了,这是怎么回事
有做空气中的邻苯二甲酸酯类分析的吗?谁有相应的分析方法? 采样方法传一个。
我们之前做液质联用的洗脱液是0.1%甲酸(ESI+和ESI-都是),现在有人提出负离子应该用甲酸铵或者乙酸铵,我想知道是什么原理呢?我不是这个领域的,麻烦大家帮帮忙,因为发文章审稿人指出了这个问题
用液相做氨基甲酸酯,单进样的时候还出峰了,过了1个小时再进样,就不出峰了。进的都是标准品。仪器是出什么问题了吗?
邻苯二甲酸酯做曲线时大家配的浓度一般是多少啊?看到有的人是1,2,4,8ppm,这样会不会太大了啊?
氨基甲酸乙酯怎么在FID是响应值那低啊,1mg/ml都不出峰,10mg/ml峰也不高,这是怎么回事[em09509]
请问有谁知道聚对苯二甲酸乙二酯(PET);乙二醇锑;间苯二甲酸;的MSDS多谢大家!
做scan时出现疑似邻苯二甲酸二乙酯的峰。但是重新进一针又没有。时有时无。请问怎么回事?样品是纯度很高的NMP(N-甲基吡咯烷酮)刚开始怀疑取样的锥形瓶的橡胶塞子被腐蚀,后来把塞子浸泡了一夜,取样上机测试没发现邻苯。
如何做PE材料中的邻苯二甲酸盐
想请教各位几个问题1.使用AccuStandard公司生产的M-606,6种酯类混标配制邻苯二甲酸二丁酯及邻苯二甲酸二(2-乙基己基)酯标准曲线,溶剂使用正己烷,外标法配置曲线,各浓度点分别为40ug/L、80ug/L、160ug/L、240ug/L、320ug/L、400ug/L。发现邻苯二甲酸二丁酯曲线相关系数大于0.990,但邻苯二甲酸二(2-乙基己基)酯则出现低浓度点峰面积高于高浓度点峰面积的情况。这是什么原因?(已通过Qedit查看过不是积分面积缺失之类的问题)2.使用容量瓶液液萃取,100mL水样加入5mL正己烷,做空白水样加标,加标后浓度应为80ug/L,但做了几只发现加标萃取后浓度仅为20ug/L或更低,回收率很差,这是什么原因?邻苯二甲酸二丁酯及邻苯二甲酸二(2-乙基己基)酯溶解不完全?液液萃取时有什么步骤是需要注意的吗?以上问题求助各位
咨询护肤品测试邻苯二甲酸酯(3P或6P)与二恶烷的问题!是检测公司的大侠们看看,给给回复。公司简介,测试费用与优惠活动等。
甲酸甲酯 甲酸乙酯 大家知道这两种物质的密度各是多少呀?
[color=#444444]邻苯二甲酸酯我们实验室以前一直是用液相做,现在老板说是要用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做节省成本,但是高沸点的几个峰,比如辛酯、乙基己酯、壬酯等都显著比前面的甲酯、乙酯小很多。而且浓度越低偏小越多,100ppm是峰高有十几,10ppm时就没峰了。用FID和ECD都出现这种情况。[/color][color=#444444]做邻苯二甲酸酯的前辈貌似比较多,希望不吝赐教,拜谢[/color][color=#444444]色谱条件:[/color][color=#444444]HP-5,30m*0.32mm*0.25um[/color][color=#444444]60℃ for 1min,20℃/min to 220℃,5℃/min to 280 ℃[/color][color=#444444]柱头压0.06MPa[/color][color=#444444]进样口温度280℃,分流比1:10[/color][color=#444444]检测器280℃[/color][color=#444444]进样器和检测器温度在250到300之间都试过,尾吹在30到80都试过[/color][color=#444444]另外还有个现象,峰大小还与初始柱温有关,初温60℃时峰面积会比80℃时大四五倍,我猜测与低温下的柱头富集效应有关,但是没想到差别会这么明显,不知道大家是否也遇到这种现象?[/color]
大家好!最近在用气相检测氨基甲酸乙酯,但是只有溶剂峰 没有标样峰 具体条件是:安捷伦6890,FID检测器,PEG-20M毛细管柱,20m*025mm*0.5μm,进样口200度,检测器250度,不分流进样 柱前压10pis。升温程序::初始柱温40%,保持0.75min,以3℃/min的速率升至90℃,再以l℃/min的速率升至110℃,保持 2.Omin,再以25℃/min的速率升至220℃,保持1.Omin。结果在2分多钟只出了一个大的峰,应该是丙酮峰,其他没有了 ,我用的是200μg/l的标样做的 ,用10mg/l的标样也试过 。而且在升温过程中 当温度快到200度的时候 基线会上升,开始基线是21pA,到达220时基线稳定100PA左右
弱弱的问一下做LC-APCI-MS,APCI+的时候,是不是也要加点甲酸好点? 是不是与ESI+时候一样的?







 我要推广仪器
我要推广仪器
 下载APP
下载APP