(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
请教各位大神:甲苯和醋酸仲丁酯混合后经检测为何只出一个峰值? 甲苯和醋酸仲丁酯单独进样出峰时间都是一样的!后将载气流速调慢,结果是一样的!请问怎样才能将这两种物质区分开来?
请教一个技术问题,我现在做酸碱滴定,用0.1N NaOH滴定醋酸乙酯中的酸度,酚酞指示终点。样品体系为:70%醋酸2%硫酸10%水15%醋酸乙酯其他,杂志据说,使用自动电位滴定仪可以一次滴出醋酸和硫酸的含量,是真的吗?PS,由于体系中有不少的酯,样品制备时不能用水稀释,否则会引起酯的水解。
用Wax柱子做丙烯酸丁酯的样品测定,死活分离不出丙酸丁酯,后来问了另一家做相同产品的公司,他们说用FFAP柱子能分离出来,我们就照做了,可是还没分离出来。搞得我现在严重怀疑是我们买的丙酸丁酯有问题,买的是国药的GCS丙酸丁酯,批号是O2789,有人遇到同样的问题吗?
大家好,我想用GC检测2甲基乙酸丁酯的左右旋异构体,试验了安捷伦的CycloSil-B手性柱,柱温40保持10分钟,1度/分升至80度。但结果左右旋异构体不能分开,仍然是一个峰。查了一些文献,都是二维气相,用串联的两根柱子来检测香精的左右旋异构体的,体系显得比较复杂。请问只用一根手性柱可以检测左右旋异构体吗?我应该怎么做呢?非常感谢。
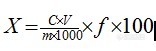
[align=center]医用胶中α-氰基丙烯酸正丁酯的测定[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]品控部:赵杨瑞[/align][b]1.原理[/b] 试样经二氯甲烷溶解定容,采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定,保留时间定性,峰面积外标法定量。实验方法参考史迎杰等[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法测定医用胶粘剂中α-氰基丙烯酸正丁酯的含量。[b]材料与方法[/b]2.1仪器设备 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:配FID检测器,分析天平:感量为0.0001g 。2.2试剂二氯甲烷:分析纯。α-氰基丙烯酸正丁酯标准品。[b]试样处理[/b]3.1 样品配制:准确称取样品50mg(精确至0.00001g),置于50mL容量瓶中,加入二氯甲烷溶解定容,摇匀。3.2[b] [/b]色谱条件:3.2.1色谱柱:(5%-苯基)-甲基聚硅氧烷为固定相的毛细管色谱柱,柱长30m,内径0.25mm,膜厚0.25μm或同种极性的色谱柱;3.2.2流速:1.0mL/min ;3.2.3进样体积1.0μL;分流比:50:1。3.2.4柱温:程序升温,初始温度120℃保持3 min,以5℃/min的速率升温至180℃,保持2 min;进样口温度:120℃;检测器:200℃。3.3外标法计算公式:[align=center][img=,156,51]http://ng1.17img.cn/bbsfiles/images/2017/09/201709110853_01_2904018_3.png[/img][/align]式中:X-样品中α-氰基丙烯酸正丁酯的含量,% C-由标准曲线所得样品溶液各组分浓度,mg/ mL; V-定容体积,mL;m-称样质量g;f-稀释倍数。 两次测试结果的相对误差小于10%即为测试平行。[b]4实验结果[/b]4.1外标法标准曲线线性的确定准确称量α-氰基丙烯酸正丁酯标准品约105mg,精密称定,置于10 mL容量瓶,加入二氯甲烷溶解定容,摇匀,配置成浓度为10.5mg/ml的标准品储备液,再精密配制成8.0mg/mL,4.0mg/mL,2.0mg/mL,1.0mg/mL的标准品溶液,置于进样小瓶中,密封。测定氰基丙烯酸正丁酯浓度与峰面积的相关性,确定相关系数及线性范围,标准曲线见图1。可见,α-氰基丙烯酸正丁酯在1.0-10.5mg/mL范围内,Y=227330X-124184,R[sup]2[/sup]=0.9976506;含量与色谱峰面积呈显著的线性关系,可满足定量分析的需要。[align=center][img=,610,423]http://ng1.17img.cn/bbsfiles/images/2017/09/201709110854_01_2904018_3.png[/img][/align][align=center] 图1 α-氰基丙烯酸正丁酯标准曲线[/align]4.2检出限取0.5mg/mL和1.0mg/mL标准溶液梯度稀释进样,至S/N=3±1,确定出α-氰基丙烯酸正丁酯的最低检出限0.5%。4.3加标回收及重复性 对样品进行加标回收实验,加标浓度设1.0mg/mL,回收率结果见图2,可见对样品进行的加标回收率在95.8%左右。对样品进行重复性实验结果见图3,结果可见,RSD为1.011%,由图2和图3结果表明本实验方法能够满足分析要求。[align=center][img=,575,101]http://ng1.17img.cn/bbsfiles/images/2017/09/201709110855_01_2904018_3.png[/img][/align][align=center]图2 α-氰基丙烯酸正丁酯样品加标回收率结果[/align][align=center][img=,586,451]http://ng1.17img.cn/bbsfiles/images/2017/09/201709110855_02_2904018_3.png[/img] [/align][align=center] 图3 α-氰基丙烯酸正丁酯重复性实验[/align][align=left][b]5.结论[/b][/align]综上所述:医用胶中α-氰基丙烯酸正丁酯的测定方法学从线性、重复性、回收率、准确度、最低检出限均符合分析要求。本方法的α-氰基丙烯酸正丁酯的检出限为0.5%,本方法可以用于医用胶中α-氰基丙烯酸正丁酯的测定。
方法GBT 18204.2-2014 公共场所卫生检验方法 第2部分 化学污染物尿素的测定中安替比林需要先用1+1硫酸溶解,然后用混酸定容,硫酸浓度不能大于1+1,想问一下混酸怎么配置?[img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901110813319903_3040_3472894_3.png[/img]
丁酸戊酯丁酸异戊酯异丁酸戊酯异丁酸异戊酯这四个从质谱上 有什么明显的区别吗? 公司没有这些原料 我没法进仪器区分 是刚才分析样品是看到的一个峰,区分不开到底是啥, 大家支支招 还是要我把谱图上传 都可以的。。。。上个附件,RT:12.577 大家有空帮忙看看 这个到底是什么??

在饮用水硫酸盐检测中,我们用到铬酸钡分光光度法(热法);其中配制铬酸钡混悬液有过失败教训,现贴出来共大家分享,不至于走弯路:GB/T5750.5-2006中描述如下:[img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001071142_194781_1615997_3.jpg[/img]我们的做法:铬酸钾和氯化钡分别溶解在大烧杯中,加热至沸,趁热倾入另一大烧杯中,让其产生沉淀([color=#DC143C]千万不能搅拌[/color])自然沉降至上层液体清亮,一般2-3小时,倾去上清液,加入纯水清洗沉淀多次,此时可用玻棒轻轻搅动液体,直到上清液中用硝酸银溶液检测不出氯离子为准,最后定容就ok了。
催化合成丁二酸二丁酯第一章 绪论1.1 概要羧酸酯是一类重要的化工原料,低级的酯一般都是水果香味,可作香料(如醋酸异戊酯有香蕉味,戊酸乙酯有苹果香味等)。液态的酯能溶解很多有机物,故常用作溶剂。有些酯还可用作塑料、橡胶的增塑剂。丁二酸二丁酯是一种新型塑料工业的增塑剂,该增塑剂为无色透明液体,常用作有机合成中间体、食物添加剂、气象色谱固定液,是一种昆虫驱避剂,用于驱除蟑螂、蚂蚁等害虫,它的合成与其它酯类化合物一样,由相应的酸和醇通过酯化反应而制得.以往的酯化反应多采用浓硫酸做催化剂,而浓硫酸有腐蚀性,使得酯化反应副反应多、后处理困难、产品色泽较差,同时,在后处理过程中还会产生大量的含硫废水污染环境.为解决浓硫酸作催化剂时的缺点,人们已研究了其它催化剂来代替浓硫酸,但对于丁二酸二丁酯的合成研究的较少,虽有人将TiO2/S042- 固体超强酸用于催化合成丁二酸二丁酯,但该催化剂的制备较为复杂,成本较高,不利于工业化生产.随着人们环保意识的增强,对于酯化反应的催化剂进行了广泛的研究,作者曾注意到结晶硫酸氢钠是一种常见的结晶无机盐,保管、运输、使用均很方便,又能克服无机酸的强腐蚀性,因此作者将研究把硫酸氢钠直接用于催化合成丁二酸二丁酯,主要研究该物质的增塑剂性能和合成该物质所使用的催化剂。
大家过年好。我最近在做链霉素,用到了七氟丁酸(离子对试剂),我想问一下,七氟丁酸在液质中的作用是什么?使用时需要注意那些事项?谢谢各位能够提供相应的资料。
【作者】 马铭研; 周丹丹; 于治国;【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 辽宁沈阳110016; 辽宁沈阳110016;【摘要】 目的:比较研究大鼠尾静脉注射与局部皮肤给予酮咯酸氨丁三醇的药动学行为。方法:采用HPLC法,色谱柱:Dia-monsil C18柱(200mm×4.6mm,5μm);流动相:甲醇-水-三乙胺-冰醋酸(80∶19.9∶0.02∶0.08);流速:1.0mL.min-1;柱温:30℃;检测波长:313nm。结果:酮咯酸氨丁三醇在0.2~100mg.L-1范围内与峰面积呈良好的线性关系(r=0.999 0),日内RSD为2.3%~5.1%,日间RSD为2.2%~12.2%,萃取回收率为86.8%~96.2%,注射剂和凝胶剂的T1/2α分别为(0.4±0.3)h,(2.9±2.6)h;T1/2β分别为(2.7±2.0)h,(9.0±8.5)h。结论:本试验建立的方法操作简单,方法灵敏、特异,结果准确。酮咯酸在大鼠体内药动学行为符合二房室模型;外用给药透皮吸收良好。【谱图】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208142206_383898_1609970_3.jpg
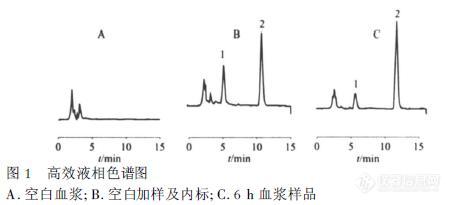
[align=center][b]盐酸雷尼替丁系统适用性试验-2015中国药典[/b][/align]色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相A:酸盐缓冲液(取磷酸6.8ML置1900ML水中,加入50%氢氧化钠溶液8.6ML,加水至2000ML,用磷酸或50%氢氧化钠溶液调节pH 值至7.1±0.05):乙腈=98:2流动相B:磷酸盐缓冲液-乙腈(78 : 22)梯度程序:[img=,617,209]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416404930_2830_2428063_3.png!w617x209.jpg[/img]流速:1.5ML/min柱温:35℃波长:230nm进样量:10μL[img=,534,227]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416589880_2883_2428063_3.png!w534x227.jpg[/img]结论:1. 出峰顺序为杂质I,雷尼替丁2. 杂质I的相对保留时间约为0.853. 雷尼替丁峰和杂质I峰的分离度大于4.0以上指标都符合药典要求。本应用来源于Kromasil微信公众号
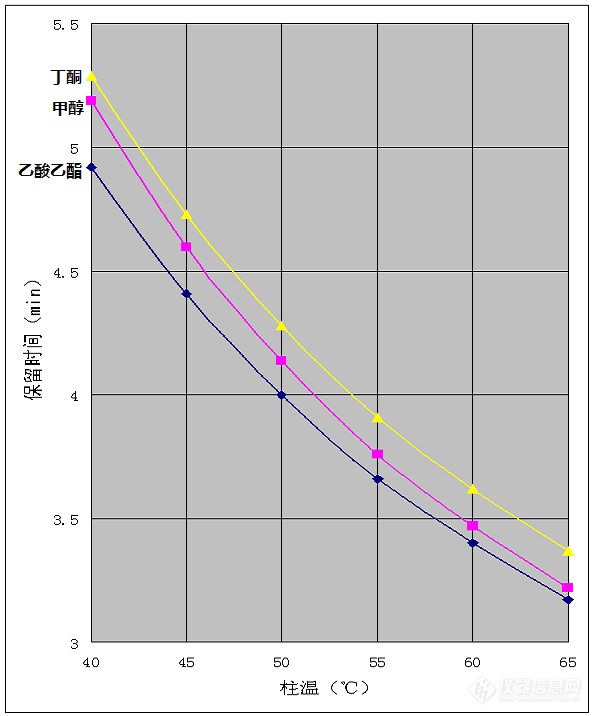
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
甲醇中乳酸乙酯不稳定,结果买了乙腈中乳酸乙酯,农残级乙腈稀释 做HJ734,乙腈中乳酸乙酯组合三热脱附进样 乳酸乙酯分解!在乳酸乙酯相邻峰位置出现分解峰,应该是氧化产物。各位用乙腈中乳酸乙酯稳定吗,是一个峰吗?
我公司做循环冷却水中的铜离子测定?根据国家标准《GBT 13689-2007 铜的测定》要用四氯化碳进行萃取!!!可四氯化碳毒性太大了,打算尝试用乙酸丁酯代替之。在这里乙酸丁酯能代替四氯化碳作萃取剂嘛?望高手解答。如有关于有机萃取剂的资料 也希望能共享下。
用乙酸丁酯萃取矿石中的金,上原子吸收火焰法测定,没有吸光度啊。为什么呢?原吸的参数都调好了呀。
各位老师,现在实验室在做氯丙醇脂肪酸酯,参照标准[font=Verdana, Arial][color=#333333][back=#f4f1e2]GB 5009.191-2016 食品安全国家标准 食品中氯丙醇及其脂肪酸酯含量的测定。其中衍生剂七氯丁酰基咪唑需要用气密针吸取,但操作过程中衍生剂容易变成白色固体,用气密针吸不起来,所以改成[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸,但好像衍生效果不好。请教各位是不能用[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸吗?为什么标准中规定要用气密针?万分感谢[img]https://simg.instrument.com.cn/bbs/images/default/em09512.gif[/img]。[/back][/color][/font]
本人实验中要分开γ-丁内酯、1,4-丁二醇和丁二酸二甲酯,但γ-丁内酯和丁二酸二甲酯可以分,加入少量的1,4-丁二醇也可以分,但一旦量大了1,4-丁二醇就显示两个峰,并且如果继续加1,4-丁二醇则又会变为一个峰,试问高手这是怎么回事呀?是不是我条件有问题呀,我用的是程序升温。非极性柱子
我想请明白的人指点一下,滴定管中酸式滴定管装酸性和氧化性的液体,碱式滴定管装碱性及还原性的液体,我如何知道配好的试剂应该用酸式还是碱式滴定管呢?如果用PH纸试,要是呈中性用哪个管呢?有没有人有个具体的方法呢?谢谢!
关于乙醇、乙酸乙酯、甲基异丁酮、甲苯、丁酸乙酯、异戊酸乙酯、乙酸-2-甲基-1-丁醇酯、己酸乙酯、异戊酸异戊酯、异丁酸异戊酯、己酸烯丙酯、紫罗兰酮、肉桂酸异丙酯13种物质的气相方法,这13种物质能否一针全部出来,用什么柱子合适,FID对它们是不是都有响应?希望各路高手路过能留下点意见和相关资料,不胜感激!
求助各位了,现在我需要用液相色谱定量分析氯乙酸和氯乙酸丁氧基乙酯的混合体系中氯乙酸的含量,选择什么紫外波长较为合适呢?200nm或者210nm可以吗?
土壤中铅镉测定电热板酸消解加酸量和温度控制测定土壤中重金属元素的总量时,常常使用各种酸或混合酸进行土壤样品的消解(即溶样)。消解的作用是:溶解固体物质、破坏土壤中的有机物、将各种形态的金属转变为同一种可测态。土壤消解原则:选用优级品酸,并采用少量多次用酸原则。消解方法一:称取土样0.5-2.00g,置于聚四氟乙烯坩埚中,加入少量水润湿。加入硝酸-盐酸(3+1)混合酸8ml,摇匀浸泡过夜。次日置于电热板上加热消解。电热板温度应调至100℃左右较低温度,至残余酸量较少时,加入2ml氢氟酸,稍调高温度继续消解。至残余酸量较少时,加入3ml高氯酸,调高温度继续消解。高氯酸消解过程中释放大量白烟,坩埚内残余酸消耗殆尽,消解土样呈半固体的滚动状态时,消解过程基本完成。冷却后用水转移至50ml容量瓶中,定容摇匀。此溶液可放入冰箱中保存。消解方法二:称取约0.5000g土样于25ml聚四氯乙烯坩中,用水润湿,加入10ml盐酸,电热板低温加热溶解2h。加入15ml硝酸继续加热,至余5ml。加入5ml氢氟酸并加热分解氧化硅及胶态硅酸盐。最后加入5ml高氯酸加热蒸至近干。再加入(1+5)硝酸1ml,加热溶解残渣,加入0.25g硝酸镧,溶解定容至25ml,同时做全程序试剂空白。消解方法三:准确称取0.1-0.3g(精确至0.0002g)试样于50ml聚四氟乙烯坩埚中,用水润湿,加入5ml盐酸,于通风橱内的电热板上低温加热,使样品初步分解蒸至2-3ml时,取下稍冷。加入5ml硝酸,4ml氢氟酸,2ml高氯酸。加盖后于电热板上中温加热1h左右。然后开盖继续加热除硅,应经常摇动坩埚。当加热至冒浓厚高氯酸白烟时,加盖,使黑色有机碳化物充分分解。待坩埚上的黑色物消失后,开盖驱赶白烟并蒸至内容物呈粘稠状。视消解情况,可再加入2ml硝酸,2ml氢氟酸,1ml高氯酸,重复上述消解过程。取下稍冷,用水冲洗坩埚盖和内壁,并加入1ml(1+5)硝酸溶液温热溶解残渣。转移至25ml容量瓶中,加入3ml5%磷酸氢二铵溶液,冷却后定容摇匀。讨论问题有:一、加入盐酸或盐酸与硝酸的混酸使样品初分解。1、单独加入盐酸然后再加硝酸好,或是加入盐酸与硝酸(3+1)的混酸好,加入酸的量和比例多少为好?2、是否要放置过夜,初分解时间多少为好?3、初分解的较低温度多少为好,是否100℃左右为好?4、盖上盖子。二、方法三硝酸、氢氟酸、高氯酸一起加好,还是分开加好?三、加入氢氟酸分解氧化硅及胶态硅酸盐。1、加酸量;2、温度;3、开盖;4、摇动。四、加入高氯酸,分解有机碳化物。1、加酸量;2、温度;3、盖上盖子。五、加热驱赶白烟并蒸至内容物呈粘稠状(半固体的滚动状态更形象)。六、(1+5)硝酸溶液温热溶解残渣,水定容。另技术方面讨论问题有:一、盐酸、硝酸、氢氟酸、高氯酸的作用分别是什么?二、盐酸、硝酸、氢氟酸、高氯酸的沸点各是多少,沸腾时的现象是什么样?三、是否有其它要注意的问题,可使消解完全且不损失,可减小引进的污染,减小试剂空白?四、与干法灰化消解方法的比较,有何优缺点?[B]请大家积极参与讨论,有积分和经验奖励,灌水将删帖!~~~[/B]——夜市(raoqun20)
我要分析发酵产物中以下几种产物:丙酮、乙醇、丁醇、乙酸和丁酸。用的是“10% Carbowax-20 M, 0.10% H3PO4,support 80/100Chromosorb WAW” 玻璃填充柱和FID检测器。 因为发酵的底物中有一定浓度的糖类物质,所以采取别人的建议在衬管(liner)中填充了石英棉以阻止糖类物质进入柱子,否则糖类物质会很快的污染衬管而出现奇怪的峰。现在发现另外一个问题:那就是乙酸和丁酸总有残留,也就是说在测完一个样后,如果再测一个水样,会出现乙酸和丁酸的峰,而且峰面积比较大。不知道是乙酸和丁酸残留在柱子中,还是liner中或者其他什么地方?是否因为温度不够高?用的方法:进样口温度 225 oC;检测器温度:225 oC;柱温箱(oven)初始温度 40 oC,ramp 1: 40 oC/min for 3min, 最终温度 200 oC for 7 min; 不知有没有人有类似经历,可以给出您的建议解决这个问题?多谢!
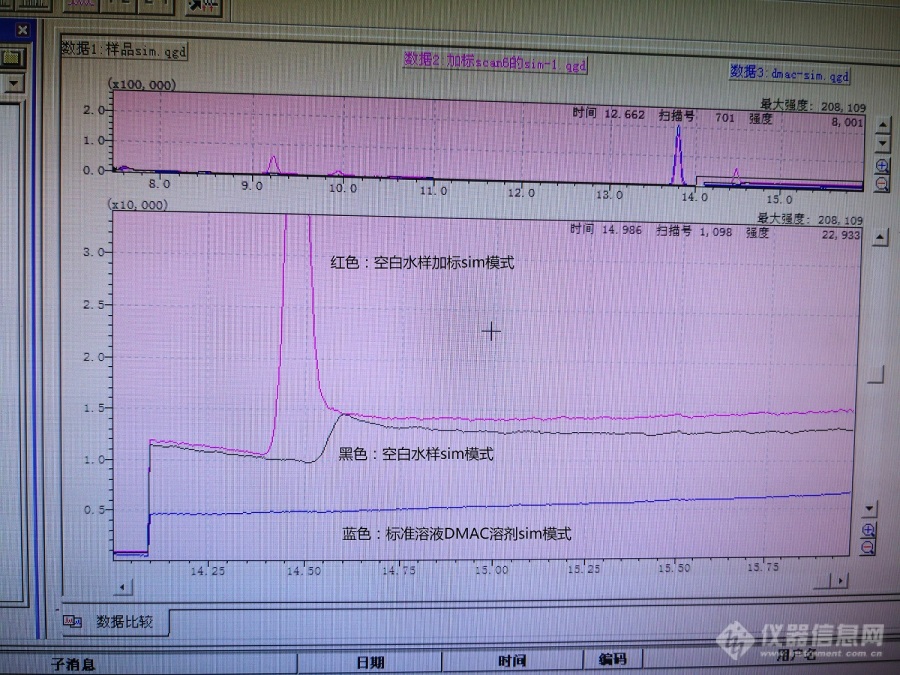
[font=宋体]用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]新建水样中乙酸丁酯等检测方法,柱子[/font]wax[font=宋体],程序升温[/font]60[font=宋体]℃([/font]8.5min[font=宋体])[/font]-5[font='微软雅黑','sans-serif']℃[/font][font='微软雅黑','sans-serif']/min-70℃-10℃/min-200℃-20℃/min-250℃[/font][font=宋体]([/font][font='微软雅黑','sans-serif']5min[/font][font=宋体]),乙酸丁酯出峰时间[/font][font='微软雅黑','sans-serif']14.6min[/font][font=宋体]。用母液(溶剂[/font][font='微软雅黑','sans-serif']DMAC[/font][font=宋体])进样峰型都很好。但是进样线性溶液(取母液用水稀释)、加标样品(母液加水样)以及溶剂水,基线就会在乙酸丁酯位置出现突越(见下图[/font][font='微软雅黑','sans-serif']14.6min[/font][font=宋体])。调整升温速率,突越总是跟乙酸丁酯一起。也老化过柱子,没有改善。请教各位老师,该突越是基线问题还是有杂质干扰,该如何去除。[/font][font=宋体][img=,656,491]https://ng1.17img.cn/bbsfiles/images/2020/03/202003261051583378_4789_2332103_3.jpg!w690x517.jpg[/img][/font]
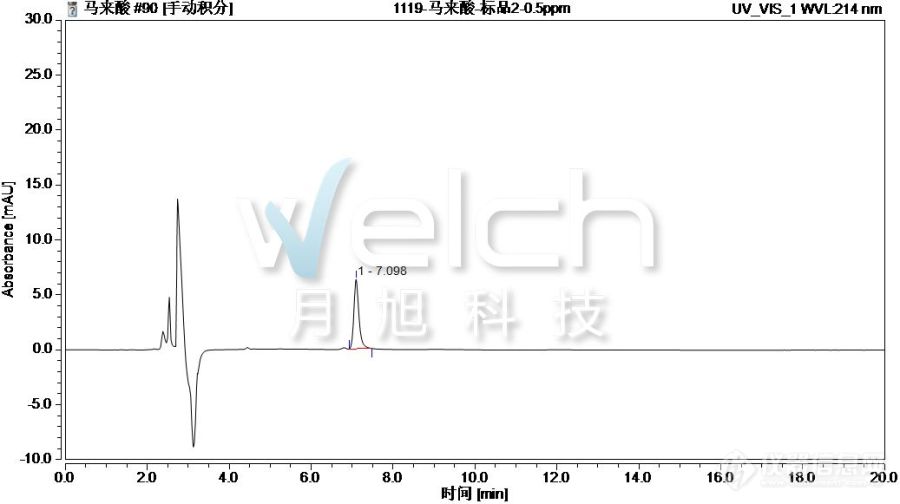
[align=center][b][img=,600,293]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520373979_4565_932_3.gif!w495x242.jpg[/img][/b][/align][b]鱼丸中的顺丁烯二酸[/b]鱼丸!一种在我们的餐桌上会经常见到的鱼肉衍生品。它没有刺,可以让你吃的更过瘾,而且呢也更利于保存,不管是拿来做汤或者涮火锅都是非常棒的选择,他的同胞兄弟还有虾滑鱼滑等等。我们今天就给大家带来我们日常的生活中很常见的一种添加剂,顺丁烯二酸也叫马来酸。它在我们的日常生活中很常见,在果汁,茶饮,运动饮料中经常会见到他的身影,用来增强特殊果香味,来增强口感。这种添加剂在其他的食品中也会出现,我们今天就一起来看一下,在如此鲜美的鱼丸中顺丁烯二酸的检测情况。[b]一-适用范围[/b]适用于鱼丸中顺丁烯二酸的检测。[b]二-提取步骤[/b]称取1g鱼丸1、用10mL 50%乙醇提取,超声10min,离心8000rpm 5min,移取上清液;2、重复(1)过程,合并上清液,用5%氨水PH调至8左右,待净化。[b]三-SPE净化步骤[/b]SPE柱:月旭WelchromSAX规格:150mg/6mL。活化:5mL 甲醇、5mL 水,弃去;上样:待净化液取4mL上样,控制流速,不宜过快,弃去;淋洗:3mL水,弃去,抽干;洗脱:5mL的0.1%硫酸,抽干,收集于离心管中,过0.22μm有机滤膜,供液相色谱仪测定。[b]四-色谱条件[/b]色谱柱:月旭Xtimate C18 4.6×250mm,5μm;流动相:A-0.1%磷酸水,B-甲醇(A:B=98:2混匀走单泵);流速:1.000mL/min;柱温:30℃;进样量:20μL;检测波长:214nm。[align=left][b]五-谱图和加标回收率结果[/b][/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520424464_8327_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center]图1.顺丁烯二酸0.5mg/L标准图谱[/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520468869_1163_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center]图2.鱼丸样过柱图谱[/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520502202_1067_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center]图3.顺丁烯二酸鱼丸样加标12.5mg/kg过柱图谱[/align][align=center][b][img=,600,335]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520540286_1398_932_3.jpg!w690x386.jpg[/img][/b][/align][align=center]图4.顺丁烯二酸鱼丸样加标50mg/kg过柱图谱[/align][align=center][b][img=,600,147]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520567559_7391_932_3.png!w650x160.jpg[/img][/b][/align][align=center]表1.顺丁烯二酸过SAX小柱加标回收表[/align][b]六-相关产品信息[/b][align=center][b][b][img=,600,317]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251520598272_2284_932_3.jpg!w690x365.jpg[/img][/b][/b][/align]
重铬酸价洗液,具有很强的毒性,一直很想找一个洗液替代,但是查询了许多资料,没找到洗脱能力与之匹敌的替代洗液,望大家分享,有能洗脱能力强且低毒性的洗液,谢谢!
[color=#444444]关于 农业部 1025号公告-18-2008 动物源性食品中β-受体激动剂残留检测液相色谱-串联质谱法 中样品的提取为什么是用乙酸乙酯和叔丁基甲醚两种溶剂来提取,它们在提取过程中的作用各是什么?[/color]
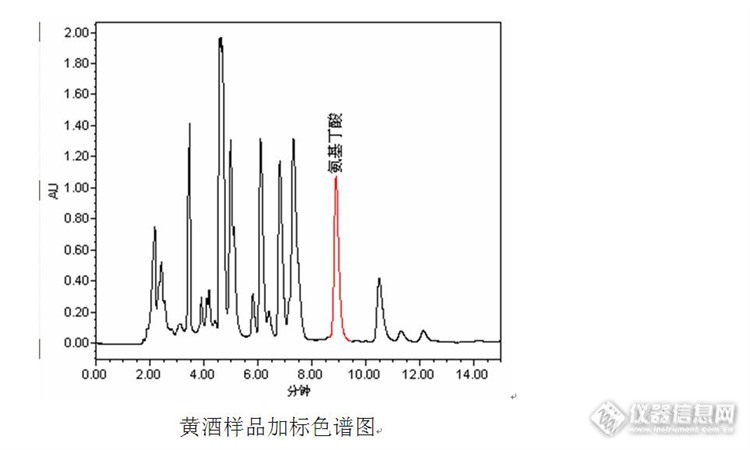
黄酒中γ-氨基丁酸含量测定的辛酸历程 近日实验室收到一批黄酒样品,该批黄酒是用发芽糙米为原料酿造而成,客户要求测定黄酒中的γ-氨基丁酸含量。由于之前实验室以丹磺酰氯为衍生试剂,建立了高效液相色谱法测定发芽糙米中γ-氨基丁酸含量的实验方法,并对实验方法的线性、精密度以及回收率进行了确认,均可以满足发芽糙米中γ-氨基丁酸含量测定要求,因此拿到黄酒样品后直接按照发芽糙米的前处理方法和色谱方法进行分析。链接如下:http://bbs.instrument.com.cn/shtml/20141226/5591256/。然而事与愿违,在测定的液相色谱图中压根就没有见到γ-氨基丁酸的色谱峰,反而在11.5min左右有个小的色谱峰,其峰高与发芽糙米中γ-氨基丁酸峰高有点相似,初步怀疑是保留时间发生了漂移,与发芽糙米样品色谱图对比后发现,在发芽糙米样品色谱图中该保留时间处也出现了一个相似的小峰,因此将该色谱峰是γ-氨基丁酸的可能性排除。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311333_530568_1669358_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412311334_530570_1669358_3.jpg 原本该实验到此结束,准备将实验结果反馈给客户:黄酒中γ-氨基丁酸的检测结果为“未检出”。为了保证数据的准确性和可靠性,在黄酒样品中进行加标实验,结果在加标的色谱图中也未在相应的保留时间出峰,而且11.5min左右的色谱峰也没有增大,因此决定先将“未检出”的结果搁置,并对实验方法进行分析。 经过对样品前处理过程和色谱方法的分析,觉得可能造成加标样品中γ-氨基丁酸未检出的原因可能有:(1)保留时间漂移。由于流动相需要调节pH值,同时样品前处理过程中也涉及到酸、碱溶液的使用,怀疑是流动相或者样品pH的改变导致保留时间的漂移,从而未在原有的保留时间出现应有的色谱峰。然而重新配制流动相和前处理样品,加标样品测定结果依然是“未检出”,对比加标和不加标样品的色谱图,两者几乎一样,也没有峰面积或峰高变化明显的色谱峰;(2)衍生试剂失效。丹磺酰氯对光和湿敏感,不稳定,放置时间久了会生产二氯亚砜并继续分解成其他物质,影响其在有机溶剂中的溶解度,也会影响结果。可是为了排除衍生试剂的问题,重新打开一瓶刚购置不久的丹磺酰氯试剂,并重新试验,结果仍然不理想;(3)衍生条件控制不当。之前用相似的方法测定牛磺酸含量以及测定发芽糙米中γ-氨基丁酸含量时曾出现过衍生过程条件控制不当造成衍生不完全或者不能衍生的情况,可是与黄酒样品同一批处理的γ-氨基丁酸标准溶液和发芽糙米样品均能衍生成功,并正常出峰,为何唯独黄酒样品不出峰呢?在百思不得其解之际,看到同事在滴定黄酒中总酸,忽然间若有所悟:黄酒中的γ-氨基丁酸需要在碱性条件下才能与丹磺酰氯发生衍生反应,而黄酒是酸性介质,pH值一般在3~5之间,同时黄酒为酿造产物,对酸碱性具有一定的缓冲能力。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311336_530572_1669358_3.jpg 通过比较发现:黄酒为酸性样品,缓冲能力较强,按照发芽糙米样品前处理方法直接加入0.5mL 碳酸钠(pH9.8)可能不能达到合适的衍生反应条件,最终导致黄酒样品中γ-氨基丁酸“未检出”。 找到问题后调整实验方案,先将黄酒样品调整至中性,然后再按照发芽糙米样品方法进行前处理。调整实验方案后,黄酒样品中γ-氨基丁酸测定的色谱图如下图。从色谱图中可以发现,经过实验方案的调整黄酒样品中检出了γ-氨基丁酸的存在。http://ng1.17img.cn/bbsfiles/images/2014/12/201412311337_530573_166
请教α-酮丁酸溶液的配置经验各位同行: 小弟我最近遇到个难题, 配α-酮丁酸溶液的时候发现这药品性状是熔融状态的,称量时用药匙一舀就沾一药匙,甩都甩不下来,而且好中的刺激性气味. 而改用其钠盐做替代物又确实划不来,价格贵了近7倍,且效果没什么太大差别! 哪位有经验的能介绍下好方法. 先谢谢了!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP