请问磷酸二氢铵、草酸亚铁的国家标准在那里可以找到?
草酸亚铁的确定,是分析草酸根还是分析铁含量,那种分析方法更有效,如何分析?
请问草酸亚铁的国家标准或行业标准那里有?
有那位化学分析高手指教,草酸亚铁中硫酸根的测定方法,急
求石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定草酸亚铁中铅的方法,不胜感谢
1、九水硝酸铝配制成饱和硝酸铝溶液怎么配制?比如配制成250ml饱和溶液需要加多少g九水硝酸铝及多少氨水?2、二水合草酸配制成10%草酸溶液怎么配制?
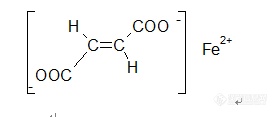
【中文名称】富马酸亚铁;反丁烯二酸亚铁【英文名称】ferrous fumarate【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/04/201204161937_361731_1855403_3.jpg 【性状】 橙红色至红棕色粉末。无臭,无味。【溶解情况】 略溶于水,微溶于乙醇。【用途】 是含铁量高的抗贫血药,用于缺铁性贫血。可用于人和动物缺铁性贫血。【制备或来源】 由反丁烯二酸经碳酸氢钠中和成盐,得到反丁烯二酸钠,再与硫酸亚铁置换而得。【包装及贮运】 桶装、内衬聚乙烯薄膜袋,遮光、密闭保存。【生产单位】 略
请问各位:在哪可买到化学纯或更好的光谱纯一水硫酸亚铁?知道的请告诉下,谢谢!
现在要分离水、乙醇、草酸二乙酯、二甲苯,假设各占1/4的含量,二甲苯不用分离异构体,准备用GDX-401或parapak P/Q作担体,PEG20M作固定液,不知道理论能不能分离?
各位大侠,小弟想请教一下,如何测定草酸二甲酯中草酸的含量。望知道的兄弟告知。谢谢
我用液相色谱仪测试甘草酸二钾流动相为30乙睛40水可以出峰但是又拖尾,然后我就加了千分之一的三氟乙酸,然后就不出峰了,我想问一下是不是吸附在柱子上了,不加三氟乙酸又可以出峰,我可不可以在加入了三氟乙酸后调高乙睛的比例就可以了。求助各位大神。
草酸测水分选用卡尔费休水分仪的时候草酸会干扰并且附着在电极上方,在测定过程吸水导致测定结果偏大;选用快速水分仪105℃测定时间5min草酸会升华。请教下各位这个草酸的水分测定是不是可以用快去水分仪降低温度去测定,又或者是使用减压干燥80℃消失的重量就是水分这种方式去测定?
本人想使用PONA柱来分析草酸二甲酯加氢的产物不知道是否可行,产物中主要就是乙二醇,草酸二甲酯,水,乙醇等。如果不行,大家有没有推荐合适的色谱柱。谢谢大家了!
水源水重铬酸钾法测量化学需氧量时 样品硫酸亚铁铵消耗量大于空白? 还有有的时候空白加入试亚铁灵 直接变成红褐色? 请问是什么原因/
[em09509]各种试剂都是新配制的。一,0.2000g硫酸亚铁样品,加30ml水,10ml硫磷混酸,2滴5g/L的二苯胺磺酸钠指示剂,此时溶液无色透明。用0.05mol/L的重铬酸钾滴定时,滴了3ml时,溶液变成绿色,随着滴定量的增加,绿色加深。滴到一定量,变成紫黑色。溶于水的硫酸亚铁,应该是绿色的吧?为什么刚开始是无色,滴加3ml的重铬酸钾后,才变成绿色的?(后来又把样品换为硫酸亚铁铵操作过程同上,现象相同!)二,作空白时,30ml水,10ml硫磷混酸,2滴二苯胺磺酸钠,用重铬酸钾滴定,滴了1ml左右,仍未变紫色,却呈浅浅的桔黄色(重铬酸钾的颜色),放置好一会,才缓慢变成紫色,随着时间延长,颜色加深,变成紫黑色。重做了几次现象一样(滴定硫酸亚铁时,变色很迅速)。不知这又是怎么回事?三,AR级的硫酸亚铁铵,含量为99.5%分子量为392.14算得亚铁含量:Fe%=55.84/392.14*0.995*100=14.17用上述方法测得结果:Fe%=13.98回收率:13.98/14.17=0.98659139回收率能不能这样算?如果可以,98.65%的回收率是好还是差?四,测螯合态铁(EDTA铁钠,商品标明的Fe含量为15%,可能含有未被螯合的铁),用上述方法测定未被螯合的铁含量,滴定1ml左右,变成紫色。算得Fe含量为0.89% 过了一会,发现紫色退去,又滴定几滴,变成紫色后,过一会,紫色又退去。如此反复。这说明什么?被螯合的铁放置一会后,也能被重铬酸钾氧化?用这种方法测未被螯合的铁含量合理吗?
请问 4%硫酸亚铁铵如何配置?是不是 4克硫酸亚铁铵加100ML水(滴几滴浓硫酸)? 我这么配完之后发现 溶液的颜色是淡黄色的。请教 高手, 4%硫酸亚铁铵如何配置
我们那边的企业所使用的净水剂,质量甚忧,许多企业所使用的净水剂都是用电屏废酸制造的,所以造成河里面有许多Zn锌和Fe铁的污染。我们去采取了一些企业的净水剂样品,用[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]做出了Zn锌、Fe铁、铅Pb的一些数据,现在碰到一个问题,如何判断它是否超标?请问各位高手,硫酸亚铁净水剂是否有判断标准?如果有,那是什么?
原子荧光测铅有可能空白荧光强度达到580(负高雅270V灯电流60mA),正常情况是空白荧光强度280。北京的专家说一般铁氰化钾含铅。回来试验,发现不是铁氰化钾的质量问题,也不是盐酸含铅(我买过一瓶高纯的进口盐酸,和我蒸馏提纯前后的盐酸含铅空白一样好。)。我改用酒石酸代替草酸实验(可以采用酒石酸、柠檬酸、邻苯二甲酸、乙酸等代替草酸实验,效果和草酸一样,参阅《中国金属学会第13届分析测试学术年会论文集》2006年P37),证实了是草酸的问题。估计草酸含铅约10mg/kg,符合试剂标准20mg/kg要求。我的铁氰化钾、盐酸、草酸都是进口的,所以不要盲目迷信进口的试剂一定好。我以前用的铁氰化钾、盐酸、草酸都是国产的质量也很好。昨天用了一瓶广州化学试剂厂的铁氰化钾,和美国进口的铁氰化钾比较,铅的空白是一样的好。所以不是盐酸含铅过高的问题。我发帖是提醒朋友们注意到草酸这种络合剂很可能含铅,这是没有人提到过的情况。
菠菜含有较多草酸,草酸不但在肠道抑制钙、铁等矿物质吸收,进入血液后还增加患肾结石的风险。所以菠菜烹调前应先焯水,草酸水溶性很强,焯水能去除大部分草酸。菠菜焯水后适合素炒、蒜炒、炒肉、炒鸡蛋、做汤、做馅、凉拌或蘸酱等各种吃法。
由于加聚铝效果不好,cod达不到排放污水管网的标准。改了净水剂,加硫酸亚铁,用石灰水调节酸碱度,昨天下午试运行的时候还好,昨天晚上没多少污水久没排,让污水在沉淀池里放这。今天早上去,一池子水都黑了。是不是水缺氧呢。
六水硫酸亚铁铵是不是易吸潮,用啥方式烘干?烘箱是多少度,60度或80度?多长时间,才能烘干,是否会把结晶水烘跑?
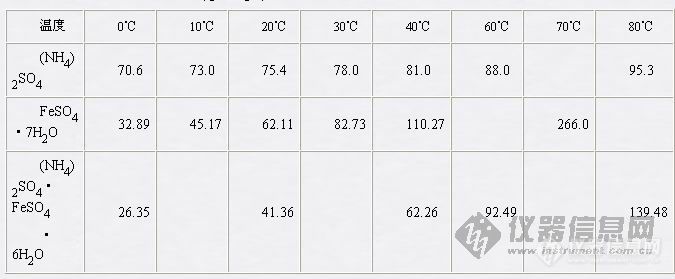
硫酸亚铁铵的制备目的原理实验目的1.了解复盐的制备方法;2.熟练过滤、蒸发、结晶等基本操作;3.了解目测比色法检验产品质量的方法。实验原理铁溶于稀硫酸中生成硫酸亚铁,并与等摩尔数的硫酸铵在水溶液中相互作用,即生成溶解度较小的浅蓝绿色硫酸亚铵FeSO4(NH4)2SO46H2O复盐晶体,反应式如下:Fe+H2SO4 = FeSO4+H2FeSO4+(NH4)2SO4+6H2O = FeSO4(NH4)2SO46H2O在空气中亚铁盐通常都易被氧化,但形成的复盐比较稳定,不易被氧化。仪器药品台称、布氏漏斗和吸滤瓶、比色管(25ml)、蒸发皿、表面皿。pH试纸、滤纸。HCl (2moldm-3)、H2SO4 (3moldm-3)、HaOH (2moldm-3)、Na2CO3(10%)、(NH4)2SO4固体。过程步骤一、铁屑表面油污的去除称取4g铁屑,放在小烧杯中,加入20ml 10% Na2CO3溶液,小火加热约10分钟,用倾析法除去碱液,用水把铁屑冲洗干净至中性,备用。二、硫酸亚铁的制备在盛有4g铁屑的小烧杯中倒入30ml 3moldm-3 H2SO4溶液,盖上表面皿,放在石棉网上用小火加热,使铁屑和H2SO4反应直至不再有气泡冒出为止 (约需20分钟)。在加热过程中应不时加入少量水,以补充被蒸发掉的水份,这样做可以防止FeSO4结晶出来。趁热减压过滤,滤液立即转移至蒸发皿中,此时溶液的pH值应在1左右。三、硫酸亚铁铵的制备根据FeSO4的理论产量,执照反应式计算所需固体硫酸铵的质量。在室温下将称出的(HN4)2SO4配制成饱和溶液加到硫酸亚铁溶液中,混合均匀,并用3moldm-3H2SO4溶液调节pH值为1-2。用小火蒸发浓缩至表面出现晶体膜为止。(蒸发过程中不宜搅动) 放置使溶液慢慢冷却,硫酸亚铁铵即可结晶出来。用减压过滤法滤出晶体,把晶体用滤纸吸干。观察晶体的形状和颜色,称出质量并计算产率。四、产品检验1.试用实验方法证明产品中含有NH4+、Fe2+和SO42-。2.Fe3+的限量分析,称取1g产品置于25ml比色管中,用15ml不含氧的蒸馏水溶解,加入2ml moldm-3HCl和1ml KNCS(1moldm-3)溶液,再加不含氧的蒸馏水至25ml刻度,摇匀后,将可呈现的红色和下列标准溶液的红色比较,确定Fe3+的含量符合哪一级的试剂的规格。标准溶液的配制在三支比色管中分别加入含有下列数量Fe3+的标准溶液各15ml (由实验室配制)(1) 含Fe3+0.5mg(符号Ⅰ级试剂)(2) 含Fe3+0.10mg(符号Ⅱ级试剂)(3) 含Fe3+0.20mg(符号Ⅲ级试剂)然后用处理试样相同的方法配制成25ml。附注:表几种化合物的溶解度数据 ( g / 100g水)[img]http://ng1.17img.cn/bbsfiles/images/2007/03/200703101024_44217_1632583_3.jpg[/img]分析思考 1.铁屑表面的油污是怎样除去的?2.为什么制备硫酸亚铁铵晶体时,溶液必须呈酸性?3.如何计算FeSO4的理论产量和反应所需(NH4)2SO4的质量?4.怎样证明产品中含有NH4+、Fe2+和SO42-离子?怎样分析产品中Fe3+的含量?
我这平时有石墨炉测铅,所以原子荧光测铅没做过,前段时间第一次做。主要参考国标5009.12第二法 和 海光用户手册,两者基本差不多,有略微差异,我主要参照海光用户手册。一开始做标准曲线,线性是很好0.9997,但是后面的点荧光值一直往上翘,数值关系,基本上是每次都是前面点荧光值的2倍再多点,导致截距在负90个荧光值左右。于是,我洗了几针,然后重测标准曲线,线性仍很好3个9,但是截距变正的170了,(原因是我忘了重新进一针标准空白,而真正的空白估计往上飘了好多,虽然是洗过好几针的),于是,我再重测标曲废了九牛二虎之力,总算把标曲勉强搞到了我的心理价位(线性0.9992,截距-30荧光值)测样,基本还可以,反正也不知道结果准不准,问题是,空白还是会有一定幅度的飘,测几个样我就校准以下空白。加标回收,因为不知道样品铅含量,所以先加了很高量的标,回收在85-90左右(因为加标量相对样品本身含量太高了,所以这个加标回收参考性估计不强吧)第一次做,只是试探下条件,载流2%盐酸,还原剂2%硼氢化钾0.5%氢氧化钠。有个疑问,国标上,铁氰化钾和草酸是分开配的,海光用户手册上是配在一起的,我试了配在一起。第一次,称了草酸、铁氰化钾,混在一块,加水,最后有些无色晶体怎么也溶不掉。第二次,我先称了草酸,配成溶液,配的浓度跟上次一样,草酸全部溶解了,再往里边加铁氰化钾,结果起白色沉淀了。是不是两者会反应?还有,加草酸是什么作用?
配亚铁溶液,用亚铁铵还是用7水的?
[size=4][font=KaiTi_GB2312]大家好,最近我在用硫酸亚铁铵滴定法测定二氧化氯的含量GB5750.11-2006上的方法。在测定过程中遇到了一些问题,我配置的试样是从市面上买的二氧化氯消毒液经活化剂活化后配置,在滴定的过程中,V1为零,因为试样中不含锰与铬;滴定V2,将配置水样倒入磷酸盐缓冲溶液与DPD混合液后溶液无颜色,滴加硫酸亚铁铵溶液颜色变红,再滴加颜色变淡,要求滴定至无色,但刚滴加至无色就迅速有红色生成,直至硫酸亚铁铵过量,我想请教一下何时为滴定终点,我配置的二氧化氯试样是否有问题?[/font][/size]
HJ828做化学需氧量,低浓度重铬酸钾的空白试验消耗硫酸亚铁铵在20ml左右,标定硫酸亚铁铵消耗的重铬酸钾在24ml左右,怀疑是水的问题,买了怡宝的纯净水,重新加热回流做了一遍,低浓度空白实验还是消耗硫酸亚铁铵20ml左右,请问问题出在哪里?谢谢!
1、硫酸亚铁铵的标定是多少ml才终点。我试了两个标定;一个是未加指示剂前,滴定25ml硫酸亚铁铵溶液(中黄色左右),加指示剂后,滴加30多ml的硫酸亚铁铵溶液。另一个则是在未加指示剂前,滴定50ml硫酸亚铁铵溶液(浅黄色),加指示剂后,滴加8ml左右的硫酸亚铁铵溶液。终点色能看得出来,但不是很亮绿。2、使用空白进行实操,滴定前没问题,终点绿色非常不明显,无法确定。煮的过程是有显非常的明显的紫色。
实验室有废弃的化学试剂,请问一般怎么处置?比如乙酸铵,草酸铵,二水合草酸,邻苯二甲酸酐,过硫酸铵,氯化铵,硫酸亚铁铵 ,氢氧化钾,柠檬酸铵,酒石酸钾钠等等,一般是中和反应掉吗?具体怎么处置的,请各位老师指导一下,多谢!
求助各位大侠用PE8000无机进样系统测试二氟草酸硼酸锂中的杂质含量的测试。
求 GB 1626 工业草酸(乙二酸)







 我要推广仪器
我要推广仪器
 下载APP
下载APP