丙酮氨基海因的 检测方法 现在条件是 波长 210 流动相 10:90 乙腈:0.003磷酸溶液 保留时间 4.2 一直有拖尾的肩峰现在流动相的极性乙腈这么大了 请问还有这么方法可以把肩峰 区分开来
各位老师好:请问,老师有检测过药物中异丙叉丙酮和二丙酮醇痕量残留的分析的经验么?请指教。谢谢
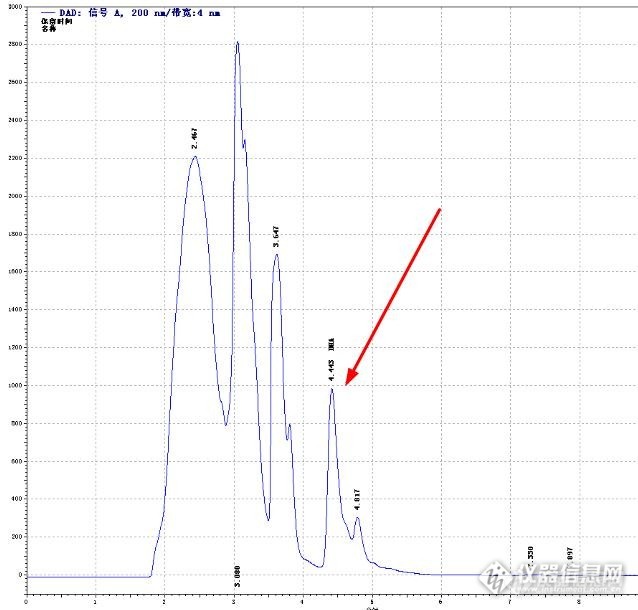
用氨基柱做二羟基丙酮(DHA),样品是合成出来的,直接进样,起初分离度非常好,可做了不到两个月分离度就不好了,究竟是什么原因呢?很纠结!流动相:乙腈:TFA(ph=3)=75:25流速:1ml/min检测器:200nm柱温:35℃正常谱图http://ng1.17img.cn/bbsfiles/images/2014/03/201403041528_491813_2563834_3.jpg非正常谱图http://ng1.17img.cn/bbsfiles/images/2014/03/201403041528_491814_2563834_3.jpg
求分离一氯丙酮、1,3-二氯丙酮、1,1-二氯丙酮、1,1,3-三氯丙酮、1,1,1-三氯丙酮、1,1,1,3-四氯丙酮的GC分析方法?
请教在甘油氧化制备二羟基丙酮的反应中使用哪种液相色谱柱分析效果较好,我们使用了氨基柱,效果不理想,求指点。
三溴丙酮和二溴丙酮要用什么色谱柱分离啊
附件是原料巯基丙酮用酒精稀释后进的gcms,请问巯基丙酮二聚体的峰到底是14.866还是22.072,或者说两者都是?还有,根据香料通则,这个东西的含量要达到95%,根据图上看有个很大的巯基丙酮,含量应该不到95%,巯基丙酮是本来就有的呢还是二聚体分解出来的?大家做原料控制的时候怎么做的呢?
做气相时柱温60℃,丙酮与二乙胺分不开,怎么办?
在做二羟基丙酮的硅烷化分析的时候,出现一个问题:当标准样品的量比较少的时候比如10mg左右的时候,气相上出来的是二羟基丙酮的硅烷化的峰,但是当标品的来那个超过20mg的时候会出来二羟基丙酮硅烷化峰以及二羟基丙酮二聚体硅烷化峰,不知哪位专家做过此物质的分析,可否指点一下。
聚乙二醇石英毛细管色谱柱对丙酮有大的作用力吗,可以把丙酮的出峰时间作为死时间吗?
做蔬菜样品时,先加的水和丙酮,加NaCl分层后,上层应该是丙酮吧?但为什么开始加的丙酮比水多很多,分层后丙酮却比水相少很多呢?而且色素全在丙酮里。 分出来的水相再加二氯甲烷提,分层后下层是二氯甲烷吗?怎么也有人说加了NaCl使二氯甲烷在水的上层呢?
请问如何定量分析?二丙酮醇分解温度较低,请问要选用什么样的溶剂作内标物呢?另外我在单独进样二丙酮醇时,一直会出现两个峰,请问如何解决?我们公司二丙酮醇的供应商拿来的检测报告上面写着纯度为99.8%,有谁知道他们是如何测出来的,而且上面的谱图是一个峰.可是我怎么做都是两个峰,请问有更好的条件分离吗?谢谢...急!~~
在做土VOC时,数据分析处理时发现丙酮,二氯甲烷和二硫化碳值都很大,主要是丙酮和二氯甲烷,有几百万那么大。做土svoc和voc时,是分房间处理的,就算交叉污染也不应该值这么大吧……纯水高我可以理解,没加内标和替代,但实验室空白和样品值那么大,我就无法理解。离子源液洗过了,再做一遍值有几万了,但还是太大。二氯甲烷可能跟我这儿的纯水机有关,我这儿用的不是屈臣氏,但丙酮那么大我就一点头绪都没有了……请问问有没有做土水气voc的大神解答一下,谢谢??![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907220838277197_6211_3862253_3.png[/img]
最近由于课题,要摸摸气相色谱,可惜是半桶水,更可悲的是GC-950。色谱柱只有不锈钢填充柱,做二甲苯跟丙酮的混合气体时候,发现出峰时间几乎一致,叠加了。真诚求教各位大侠,能想出个解决办法(除了换色谱柱),能分别测试二甲苯跟丙酮。
1丙酮不出峰2做的环境空气中丙酮,多大标曲合适3丙酮出峰在二硫化碳之前吗
请问1,2-丙二醇单甲醚和甲氧基丙酮混合物中甲氧基丙酮的含量如何分析?谢谢!
求乙酰肼,恶二唑酮,唑丙酮,三嗪酰胺检测方法,跪求,谢谢
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做二硫化碳中丙酮含量,丙酮峰分裂成俩个连续的大小差不多的峰是什么原因,在另外一台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]上就没问题,色谱柱、检测器、仪器条件都相同
W46.jpg 二硫化碳中丙酮,用填充柱分离好的,就是丙酮有点拖尾,怎么办柱温60进样口120检测器160,天美7890

我公司今年五月份进货1公斤二聚巯基丙酮(CAS55704-78-4,FEMA3450)。当时货品的状态是白色结晶,但是昨天车间使用该原料时发现状态变成了下图的样子:http://ng1.17img.cn/bbsfiles/images/2016/10/201610260946_615125_1622710_3.jpg变成了液体,有大量油珠状分层现象。我想请教一下版友,是否遇到过这种现象?我把这个原料做了色谱质谱分析,谱图见附件。RT17之后的成分都是什么呢?有网友认识吗?
溶液里还有一些四溴丙酮 需要用什么色谱柱检测出来啊?条件是什么啊
实验急需要环氧乙烷色谱纯,我用过安瓿瓶装的100ml/支的,其他规格的也可以,一支的量不能太少了,因为它很容易挥发。还需要二硫化碳中丙酮标准物质,丙酮的浓度大约为1600ug/ml,每支的体积最好大些
[color=#333333]今天做二硫化碳中丙酮,不出峰,,不知道咋整。有人会做吗??求指教[/color]
请问: 二硫化碳与丙酮反应吗?为什么用气相色谱检测重复性很差?
要测吡啶\丙酮\乙醇\DMF\乙二醇应选哪种型号色谱柱?
用气相测丙酮时,丙酮本来是一个峰的,但它分成连着的几个峰出,为什么啊?
前段时间我们用NPD检测器,HP-5色谱柱,测有机磷样品,当时想用丙酮做溶剂,有的老师提出丙酮对色谱柱的影响太大,不让用丙酮,后来选择的二氯甲烷,虽然二氯甲烷对NPD也有影响,但我们还是这样做下来了。我只是想在此请教下专家,丙酮对柱子的影响大吗?以后都要尽量避免用丙酮吗?
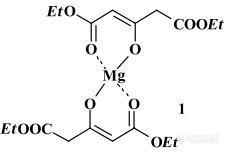
因实验需要,本人需要购买少量"双丙酮二羧酸化镁"英文名称:Mg diacetonedicarboxylate英文简写(DADC)2Mg,如有消息,请各位朋友告知小弟,感激不尽
DB-624柱,柱箱:40℃(5min)-20℃/min-190℃(10min),进样口200℃,检测器240℃,顶空进样,二甲基亚砜做溶剂,[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]顶空检测丙酮,溶剂中丙酮位置有干扰峰,进空气没有,换了不同品牌的二甲基亚砜,仍有,怎么破
各位大虾好,小弟想请教一个问题。我的试剂溶于丙酮,但是用氘代丙酮做溶剂却发现一维氢谱图中未见活泼氢的吸收峰。譬如羟基氢和氨基氢的吸收就没有体现,但是苯环上的氢和甲氧基的氢有明显的吸收。不知道这种现象是否正常,该如何解释呢?谢谢,望大家不吝赐教。







 我要推广仪器
我要推广仪器
 下载APP
下载APP