用二硫化碳来稀释甲醇中的苯,稀释时却发现溶液分层?问了一个同事,说是因为CS2中含有用来液封的水。但是水水可以溶解于甲醇里的啊?另:我在CS2原试剂瓶中也没有看到分层。奇怪?另:加热后,二硫化碳稀释甲醇中的苯溶液又不会分层,静置后又分层?
大家好,请问对二硝基苯和邻二硝基苯的纯物质可以溶于甲醇吗?请问有谁试过,主要是想配混标,其中间二硝基苯买的标液是甲醇中的标液。
用邻苯二甲醛做氨基酸的衍生试剂,结果发现甲醇溶解的邻苯二甲醛比乙醇溶解的二甲醛衍生效果要好。请问谁能解释一下这个现象吗?
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]苯。[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]新手。之前做的一直都是二硫化碳中的苯系物,现在来了一个甲醇中的苯盲样,然后做了甲醇中的苯系物标曲,只有6个峰,没有苯的峰,甲醇溶剂峰太大可能把苯的峰给遮蔽了。我现在准备用二硫化碳萃取甲醇中苯,请教各位老师这种方法可行么?值的大小影响大么?
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的谱图突然比以前宽二倍原因分析 求高手指点呀?用甲醇做溶剂测苯时,甲醇是平头峰,而且好宽,把苯峰都“举”起来了,甲醇峰拖尾好严重,各位高手,我是新手,多多指教啊~

本人一个月前评审时做过一个活性炭管中苯的考核,曲线用的是二硫化碳中的苯,当时没有二硫化碳的苯质控样,只有一个甲醇中的九种VOC100ppm(直接进样),得到结果是113,质控样没做对,也没办法只有硬着头皮报,结果碳管还是对了。今天没事在网上搜到了专家wazcq的一个帖子,http://bbs.instrument.com.cn/topic/5006342,他的帖子内容大概是说,同等浓度,同等仪器条件的情况下,甲醇中三氯苯的峰高比二硫化碳中三氯苯低(不知这样理解是否正确)。再看看我做的,同等浓度,同等仪器条件的情况下甲醇中苯的峰高比二硫化碳中苯高。我用其他仪器试一下看看,评审时用岛津GCMS做的,今天就不开了,我打开了另外两台:安捷伦7820和6890(当时还未安装)。我的试验是这样的:一瓶浓度大概在1000ppm左右的二硫化碳中苯系物(只有甲苯,乙苯,见二甲苯,应该是别人单标混的),分别用微量注射针取相同体积的液体到1ml的甲醇和1ml的二硫化碳中,注明样品A和样品B吧,分别用两台仪器进样。多次稀释的结果如下:http://ng1.17img.cn/bbsfiles/images/2015/11/201511242138_574974_1839381_3.png7820和GCMS仪器上有曲线,所以7820的的数值单位是浓度,而6890没曲线,那么6980的数值是峰面积。从图上看6890的结果与GCMS一致,同等浓度,同等仪器条件的情况下甲醇中苯系物的峰面积比二硫化碳中的大,而且比例一致;7820与专家的GC得出结果一致,同等浓度,同等仪器条件的情况下甲醇中苯系物的峰面积比二硫化碳中的小(专家的是三氯苯),在算下比例108.6/93.9=1.16与7820也可以说一致。而且调整分流比,面积比例不变。这是为什么呢?我同事说,可能是偶然。另:仪器都是自动进样,分流比都是20:1,岛津GCMS和7820柱子是HP5,6890是innowax。6890和7820都是分流衬管,岛津GCMS是不分流衬管
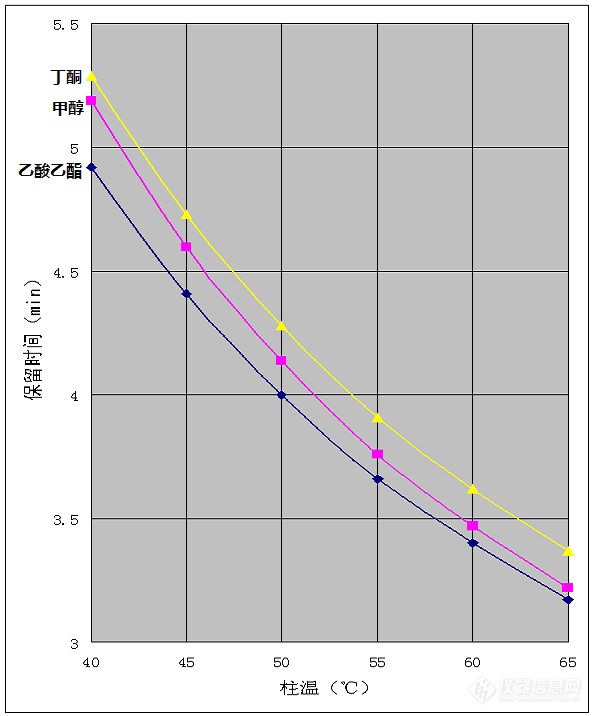
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
现在在做tvoc中的甲苯和二甲苯的能力验证,拿到的质控样是以甲醇为介质的,咱们平常做的曲线是以二硫化碳为介质的,对测出的数据不是很相信,同志们有没有买过相关的标样,把浓度贴上来参考参考。非常感谢。
[em61] 各位论友,帮帮忙!我现有一样品,其中主要分析乙二醇与苯甲醇,现求助大家帮我确定以下色谱柱以及分析条件? 非常感谢!
马来酸二甲酯和苯甲醇这两者的化学分子式是什么在高温下,又会产生什么?
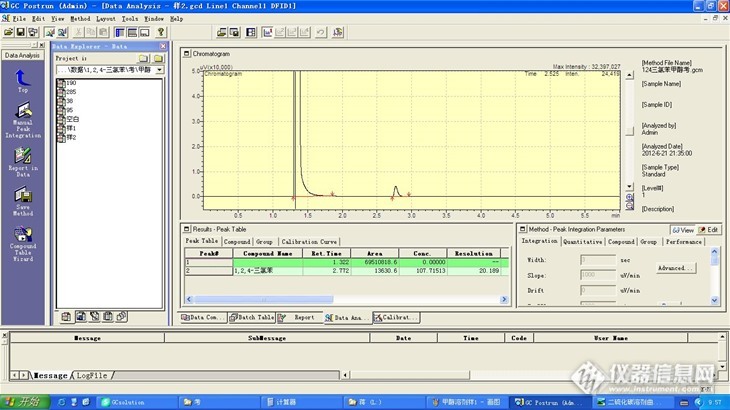
经常在论坛看见有人发帖问同一种物质在不同溶剂(讨论最多的是二硫化碳和甲醇)做标准曲线对分析结果的影响。我就以上次专家来考核甲醇中1,2,4-三氯苯盲样为例讨论下分流进样时,分别用二硫化碳和甲醇做溶剂对峰面积、保留时间的影响。 公司扩项中有环境空气和废气中1,2,4-三氯苯。方法是大气固定污染源 氯苯类化合物的测定 气相色谱法 HJ/T66-2001,专家来考核的时候带来了甲醇中1,2,4-三氯苯盲样。于是问题来了:标准里是用二硫化碳做溶剂的而盲样是以甲醇做溶剂,能用二硫化碳溶剂做的曲线来标定盲样吗?我先后用二硫化碳和甲醇做溶剂来试验。 实验部分仪器和试剂岛津气相2014C色谱仪带AOC20i自动进样器,FID检测器。1,2,4-三氯苯(色谱纯)二硫化碳(色谱纯)甲醇(色谱纯) 色谱条件毛细柱OV-101 25m*0.20mm*0.25um柱箱温度140℃,进样口温度250℃,FID检测器温度250℃,氢气40ml/min,空气400ml/min,氮气30cm/s;进样量1ul,分流比50。盲样注明考核浓度范围为1.0-150.0mg/L所以配制二硫化碳中1,2,4-三氯苯浓度分别为(ug/ml) 0,20,40,80,100,200 甲醇中1,2,4-三氯苯浓度分别为(ug/ml) 0,38,95,190,285http://ng1.17img.cn/bbsfiles/images/2013/10/201310121116_470549_2103464_3.jpg这是二硫化碳溶剂空白http://ng1.17img.cn/bbsfiles/images/2013/10/201310121116_470550_2103464_3.jpg这是甲醇溶剂空白,有拖尾。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121118_470551_2103464_3.jpg这是二硫化碳做溶剂标液100ug/mlhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121121_470553_2103464_3.jpg这是甲醇做溶剂标液95ug/mlhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121122_470554_2103464_3.jpg这是二硫化碳做溶剂的曲线http://ng1.17img.cn/bbsfiles/images/2013/10/201310121123_470555_2103464_3.jpg这是甲醇做溶剂的曲线,第一个点空白为零但是空白的数据方框不能选中(谁能解释下?这可能是不强制过零点所致),这样其实和没带入空白的曲线是一样的。有没代入空白的零点计算对截距有点影响。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121132_470559_2103464_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121133_470560_2103464_3.jpg这是盲样做的两针平行样加载二硫化碳溶剂曲线结果分别为 94.0ug/ml ,92.6ug/ml 平均值93.3ug/ml。这个结果偏小不合格。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121138_470561_2103464_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121138_470562_2103464_3.jpg这是盲样做的两针平行样加载甲醇溶剂曲线结果分别为 109.4ug/ml ,107.7ug/ml 平均值108.6ug/ml。这个结果是合格的。再看下保留时间:二硫化碳做溶剂时1,2,4-三氯苯保留时间为2.783 ,甲醇做溶剂时1,2,4-三氯苯保留时间为2.772,几乎一样。结论:用二硫化碳做溶剂1,2,4-三氯苯的峰面积明显大于以甲醇做溶剂时的峰面积,所以以二硫化碳做溶剂的曲线来测以甲醇做溶剂的样品结果会偏小。这可能和不同溶剂在气化室状态不同有关,而保留时间几乎不变。
[table=100%][tr][td]我用HPLC测对苯二酚的甲醇溶液,在2-3保留时间内出现两三个峰,而且是肩峰,没有分开。但在同条件下用对苯二酚水溶液做就只有2.8左右的一个峰,而且峰型非常好。对苯二酚纯度为98%,甲醇是色谱纯的,分析条件是流动相:甲醇:水=20:80,波长:290nm,流速1ml/min。求高人指点!![/td][/tr][/table]
做空气样中,一般用二硫化碳解析,那么以甲醇中对二甲苯与二硫化碳中对二甲苯为标准有区别吗?区别在哪里?
有谁做过3,5-二羟基苯甲醇,能够告诉我详细的操作吗?越具体越好。我是新手,望各位老师帮助。
我用的Agilent的7890GC,做的是甲醇中7种苯系物的含量测定,分别为苯、甲苯、乙苯、对二甲苯、间二甲苯、邻二甲苯、苯乙烯。柱子是用的瓦里安的52CB的毛细管柱,30m*0.32mm*0.45um,为了把对间二甲苯分开,设了个升温程序,50度保持4分钟,然后升温到80度保持至结束,载气流速2ml/min,分流比设的50。之前也试过别的条件,发现甲醇的溶剂峰非常大,苯的出峰总是在甲醇的溶剂峰的拖尾上,可能给定量带来较大误差,请问这个怎么解决?(换过手头的Waxetr柱,分对间二甲苯效果还不如这个柱,别的柱子更不行)
我目前是个实习生,要做毕业课题,是空气中苯系物的测定。现在是用甲醇做标样,甲醇中苯系物(332408)浓度为237ppm.做曲线是把甲醇稀释成5个浓度点做的。现在已经把苯。甲苯。乙苯。对间二甲苯。邻二甲苯。异丙苯的标准曲线做出来了。我做的标线浓度是10,30,50,70,90ug/ml。可是师傅要求我做方法检出限,我一窍不通,请大家帮帮忙,教我一下。最好说清楚整个过程
挥发性有机物测定,甲醇中苯系物都是7种物质,没有异丙苯,只有二硫化碳种苯系物有8种的标准溶液,大家都买的甲醇还是二硫化碳的呢?
求SN/T 2405-2009玩具中甲醇、甲苯、乙苯、二甲苯和环己酮迁移量的测定
分离甲醇做溶剂中苯 甲苯 乙苯 二甲苯(邻间对)苯乙烯都有哪些色谱柱可以分开主要是甲醇中苯分不开,甲醇峰太大,影响苯的出峰,我不能换溶剂,只能用甲醇
做苯系物的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,溶剂为甲醇,可是甲醇峰太大,覆盖了苯峰,想用二硫化碳稀释,可是二硫化碳和甲醇不互溶怎么办呀?
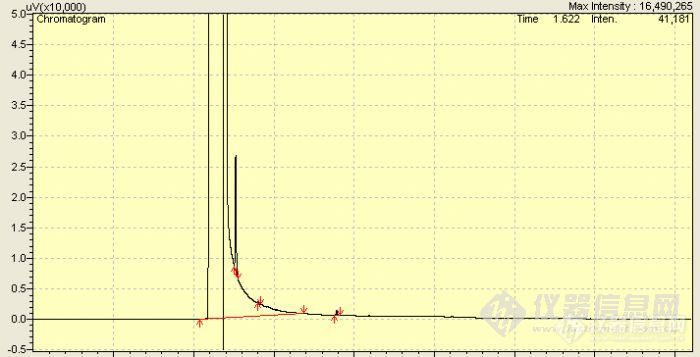
大家好!最近买了一根CD-WAX柱子分析甲醇中的苯系物,发现对间二甲苯可以很好的分离,但是甲醇的拖尾峰太大,苯峰在甲醇的拖尾峰上,而且和甲醇中杂质出峰时间相近,头疼,相比其他组分的峰,苯峰响应也很小,请问这是什么原因?该如何解决呢?请问大家在测试空气和水质中苯系物时,都是如何配置标准曲线的呢?我是用甲醇中TVOC标准溶液,经过逐级稀释配置的,然后样品是用二硫化碳萃取或解吸的,请问这样做影响大吗?因为最近一次的盲样考核数据偏大了一些。请问哪里可以买到二硫化碳中苯系物混合标准溶液呢?分析条件如下: 仪器:岛津GC-2010plus;柱子:CD-WAX 30*0.25*0.25;进样口:200;FID:250; 程序升温:初温45,保持1min,以5℃/min升温至80℃,保持1min,以15℃/min升温至110℃.http://ng1.17img.cn/bbsfiles/images/2012/03/201203091334_353536_2193530_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/03/201203091334_353537_2193530_3.jpg
邻苯混标,溶剂是二氯甲烷和正己烷,由于混标的溶剂容易挥发,稀释的时候我想用甲醇,可以吗?其实知道甲醇不合适,极性强但是又不知道该用什么稀释,求大神帮忙,谢谢了。
马来酸二甲酯和苯甲醇在反应时颜色变黄,不知道是什么原因阿,有专家吗
把甲醇中的苯注入到水中,用二硫化碳萃取,能否很好地萃取完苯?
甲醇跟,甲苯跟二甲苯可以用同一根极性柱做吗,用哪个型号的柱子效果更好一点?
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。
1、怎么用二硫化碳提取甲醇中的苯系物混合?2、怎么用正己烷提取甲醇中的有机物混合?这个问题已经困扰我几年了,谢谢各位大侠们指导.
申请能力验证发的盲样为甲醇中苯系物!但是我这里认证方法为二硫化碳的方法!问了组织单位说不影响!问厂家工程师说出峰会不一样!想请教一下大家用二硫化碳方法做甲醇中苯系物这个问题!
各位大神,帮帮忙吧,做苯系物的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测,标品是溶在甲醇里的,但甲醇峰拖尾太严重,把苯峰覆盖了,想用二硫化碳来稀释标品,但是两者不互溶呀?怎么办呀怎么办?
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。







 我要推广仪器
我要推广仪器
 下载APP
下载APP