雌二醇(17β-Estradiol)是天然雌激素的重要成分,为防止儿童性早熟等问题,食品中尤其是畜禽产品的雌二醇检测不容忽视。胶体金免疫层析技术(GICA)在检测小分子有机物方面越来越受到重视。其基本原理是胶体金颗粒能够稳定吸附蛋白质,而蛋白质的生物活性无明显变化,所以它可以取代酶标记抗体〔1,2〕。本实验采用标记有雌二醇单克隆抗体的胶体金颗粒与相对应的固定在硝酸纤维膜上的雌二醇结合物相结合,固定有雌二醇结合物的检测线处就显现明显的颜色〔3,4〕。本法具有灵敏度高、特异性强、简便快速、成本较低、结果轻易判读等优点,适用于样品的初步筛选。[b]1材料与方法[/b]11试剂与仪器氯金酸(上海化学试剂厂);牛血清白蛋白、卵清白蛋白、兔抗鼠多抗(天津联星生物公司);雌二醇标准品、雌二醇单抗、17β-Estradiol-6-one6-(o-carboxymethvyoxime)美国Sigma公司);其他试剂均为国产分析纯。硝酸纤维膜、玻璃纤维膜(美国Millipore公司);BeckmanDu530分光光度计(德国Beckman公司);低温高速离心机(军事医学科学院);点膜机(美国Bio-Dot公司)。12方法121胶体金的制备用三蒸水溶解氯金酸,使其终浓度为01g/L。先进行沸水浴,待氯金酸溶液煮沸后,每100ml加入1%柠檬酸三钠25ml,再沸水浴下快速搅拌,直到氯金酸溶液的颜色稳定,继续沸水浴10min〔5〕,冷却至室温后,用透射电镜镜检并用分光光度计检测颗粒均匀度及粒度。最后置于4℃冰箱保存备用。122最适标记蛋白量的确定用02mol/LK2CO3将胶体金溶液调至pH为82,然后取试管9支,分别加入10ml胶体金溶液。将雌二醇抗体逐级稀释后,各取等体积稀释液顺序加入上述试管中,混匀,另设一不加抗体的对照管。放置10min,在各管中加入01ml的10%NaCl,混匀后静置2h。观察各管中胶体金溶液变化,未加蛋白及加入量不足的溶液颜色由红变蓝,而加入蛋白量达到或超过时的溶液则保持不变。标记蛋白量即为最低稳定胶体金溶液不变色的蛋白量基础上再加20%。123胶体金探针的标记将抗体用0005molNaCl溶液透析过夜,离心除去蛋白沉淀,调至05mg/ml。取胶体金100ml,用0,2mol/LK2CO3将胶体金溶液调至pH为90,磁力快速搅拌下缓慢加入24ml稀释的雌二醇抗体,继续搅拌10min,加入牛血清白蛋白(BSA),使其最终浓度为1%,再搅拌10min。将初步制得的胶体金探针以4000r/mm离心20min;弃沉淀,上清以10000r/min离心60min;弃上清,沉淀用001mol/L的磷酸盐缓冲液(PBS)(含1%牛血清白蛋白)重新悬浮;同前离心洗涤2次,将沉淀用001mol/L的PBS(pH82,含1%BSA,002%NaN3)悬浮,4℃保存备用。124抗原的合成小分子物质难以固定在硝酸纤维膜上,只有使它连上大分子物质才能较好地固定。本实验采用混合酸酐法制备完全抗原。雌二醇-6-肟43mg,加入三正丁胺50μl,二氧六环4ml,冰浴冷却到10℃以下,加入氯甲酸乙酯15μl〔6〕。4~10℃下反应30min。配制卵清白蛋白(OVA)溶液(OVA10mg,3ml水,3ml二氧六环,1mol/L氢氧化钠03ml)。将配制好的OVA溶液加入反应液,4℃冰浴搅拌6h,反应过程中,用氢氧化钠维持pH值为8。反应完毕后,将反应液加入透析袋,透析2d。将透析液调至pH45,冰箱4℃,放置4天,有黄色沉淀出现。离心收集沉淀,冷冻干燥,4℃冰箱保存。将OVA溶于透析液内作为参比,用紫外光谱法对雌二醇-6肟-OVA进行定性检验。经紫外测定证实蛋白质已与雌二醇衍生物结合。125胶体金免疫层析试纸条制备测试条由玻璃纤维膜、硝酸纤维膜、加样纸、吸水纸4部分组成。将玻璃纤维膜裁成6mm的细条,然后放入含1%BSA、1%Tween-20的PB液中浸泡30min,37℃烘干,最后将胶体金探针灌注已处理好的玻璃纤维膜上,真空干燥备用。在硝酸纤维膜上用点膜机将上述抗原和兔抗鼠IgG喷成2条线,分别为检测线和对照线,经真空干燥后,用1%BSA、001mol/LPBS(pH90)封闭2h,以001mol/LPBS洗涤,再真空干燥。将30mm吸水纸、25mm硝酸纤维膜、6mm玻璃纤维膜、15mm加样纸,由顶部依次粘于PVC板上,裁成细条备用。126检测与判读样品:含已知浓度雌二醇样品的乙醇溶液分为3组,第1组不加雌二醇,第2组为02μg/ml雌二醇,第3组为04μg/ml雌二醇。将加样纸一端插入待测液,湿润后取出,水平放置,约3~5min,观察结果。如试纸条硝酸纤维膜上仅对照线呈一条紫红色带为阳性;如出现2条紫红色带为阴性;如质控线不出现紫红色带,无论检测线是否出现,测试结果均无效。[b]2结果与分析[/b]21胶体金颗粒大小选择胶体金颗粒吸附蛋白并显示检测结果,其大小是重要影响因素之一。胶体金颗粒的大小,由制备胶体金时加入的柠檬酸三钠量决定,当柠檬酸三钠加入越多,胶体金颗粒也越多,但颗粒直径越小,反之胶体金颗粒越大。胶体金颗粒小,结合蛋白量少,反应结合率低,另外难以显示明亮清楚的颜色,影响显色效果;胶体金颗粒大,其结合蛋白后不稳定,不易保存,另外其结合蛋白后难以通过膜〔7〕。本实验选用的胶体金颗粒直径约为15min,既可以通过膜,反应彻底,又易保存,反应结果显色清楚。22胶体金颗粒鉴定胶体金颗粒大小及均一程度是检测结果可靠的基础,制备完毕后须经质量鉴定才能应用。用BeckmanDu530分光光度计对胶体金进行扫描,发现其主峰宽度较小,表明制备的胶体金颗粒较均匀,λmax为520nm(图1),表明其粒度约为15nm〔8〕。用电镜观测结果(图2)与分光光度计测定结果一致。电镜观测是鉴定胶体金颗粒粒径及分布的标准,但这种方法不便用于日常工作使用,而用分光光度计测定操作简便,更适合实验室日常使用。图1胶体金的可见光谱图(略)23最适蛋白量阴性对照为未加抗体管,1~7管抗体浓度分别为10,12,14,16,18,20,22μg/ml,阳性对照为胶体金溶液。可见,本实验最适标记蛋白量为20μg/ml(图3)。24E2-OVA浓度用PBS(pH74)溶解E2-OVA浓度为005~10mg/ml,喷在硝酸纤维膜上作检测线,发现E2-OVA浓度越高,检测液中也需要越高浓度的E2与之竞争,检测灵敏度也就越低。但是,E2-OVA浓度低于05mg/ml,检测线颜色太弱,难用肉眼分辨。图2胶体金电镜图(略)阴性对照为未加抗体管;阳性对照为胶体金溶液图3胶体金与雌二醇抗体结合浓度确定实验(略)25检测限本实验的检测限是以完全看不见检测线为最低检测限〔9〕。将雌二醇标准品用甲醇配制成04,02,01,005μg/ml4个标准溶液,用上述检测方法进行检测(图4)。由图可见,最低检测线为100ng/ml。图4试纸条检测样品(略)26对比实验取30份样品,检测分为3组,第1组不加雌二醇,第2组加02μg/ml雌二醇,第3组加04μg/ml雌二醇,分别用GICA法与[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法(GC/MS)检测。实验结果显示,第1组2种方法均未检出;第2组GICA法阳性9粒,阴性1粒,GC/MS法阳性10粒;第3组2种方法均检测阳性。由此可见,与标准检测方法相比,GICA法准确度为9667%。27交叉反应试验实验结果显示,与己烯雌酚、炔雌酚、壬基酚、阿特拉津及双酚A等环境内分泌干扰物中的环境雌激素无交叉反应,表明该方法具有良好的特异性。[b]3小结[/b]目前,关于雌二醇的检测有气、液相色谱和光谱、质谱等方法,但其仪器昂贵、操作复杂、检测时间较长。本文建立的免疫胶体金层析法具有灵敏度高、特异性强、简便快速、成本较低、结果轻易判读等优点,且无需任何仪器,操作简单,适用于样品的初步筛选。[list][/list]
请问雌二醇的稳定性怎样,大家是怎样储存标准品储备液的
[table=100%][tr][td=2,1,650][font=宋体][/font][align=center][b][size=5]产品概要[/size][/b][/align][size=5] 雌二醇[b](β-Estradiol)[/b]是应用广泛的甾类同化激素,被大量用于促进反刍动物的生长。但由于雌二醇存在明显的致癌性,欧美等国家已相继禁止或严格禁止使用。我国农业部第235号文件中规定,动物源性食品中雌二醇的残留限量为不得检出。由于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](GC)分析法样本前处理及测定操作繁琐且费用高,使推广应用受到限制。[/size][align=center][b][size=5]产品用途[/size][/b][/align][size=5] 可定性、定量检测动物组织(肌肉、肝脏、虾、鱼等)、尿液、饲料、奶粉等样本中雌二醇的残留量。[/size][align=center][b][size=5]产品性能参数[/size][/b][/align][size=5] [/size][table][tr][td][size=5]产品规格[/size][/td][td][size=5]96T[/size][/td][/tr][tr][td][size=5]适用范围[/size][/td][td][size=5]动物组织(肌肉、肝脏、虾、鱼等)、尿液和饲料,奶粉等[/size][/td][/tr][tr][td][size=5]检测时间[/size][/td][td][size=5]1.5个小时[/size][/td][/tr][tr][td][size=5]检测下限[/size][/td][td][size=5]鸡肉/肝、猪肉/肝、虾、鱼 :5ppb饲料 :80ppb尿液 :2.5ppb[/size][/td][/tr][tr][td][size=5]灵敏度[/size][/td][td][size=5]0.1ppb[/size][/td][/tr][tr][td][size=5]前处理流程[/size][/td][td][size=5]组织:均质、振荡离心、吹干、溶解、反萃取、振荡离心、吹干、复溶[/size][/td][/tr][tr][td][size=5]交叉反应率[/size][/td][td][size=5]雌 二 醇 :100%雌二醇-3甲醚:30%炔 雌 醇 :7%雌酚酮 :小于0.1%苯甲酸雌二醇 :小于0.1%雌 三 醇 :小于0.1%[/size][/td][/tr][tr][td][size=5]结果判定[/size][/td][td][size=5]定性:根据样本孔颜色与标准品孔颜色进行比对定量:运用酶标仪进行计算[/size][/td][/tr][/table][/td][/tr][tr][td=2,1][size=5][i][b]无锡康纳生物科技有限公司 0510-85191271,13771068995,杨小姐[/b][/i][/size][/td][/tr][/table]
各位大侠:小弟目前正在做四环素和雌二醇的共分离实验,但两种物质各自的最优分离条件都无法分离出另一种物质,即完全无信号响应。不知我这同一色谱条件分离此二种物质的目标能否实现?目前我使用的四环素色谱条件:流动相为甲醇-缓冲液(0.1 mol/L丙二酸+0.05 mol/L氯化镁,用氨水将 pH值调至 6.5)(50:50, v/v),流速为1.0 mL/min。采用荧光检测,激发波长为 375 nm,测定波长为 535 nm。雌二醇色谱条件:流动相为乙腈-水(55:45,v/v)。采用紫外检测,波长为205nm。色谱柱为C18,25cm柱。谢谢各位!
我试过好几种反相柱,α β雌二醇分不开,有做过的老师吗?请指教!
大家有没有测过雌二醇,雌酮等物质啊 谢谢
[table=100%][tr][td][url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]17a-雌二醇和17b-雌三醇分不开,流动相我用了乙腈和千分之0.25的氨水,但是这两个激素就是分不开,大家有没有什么好的建议?[/td][/tr][/table]
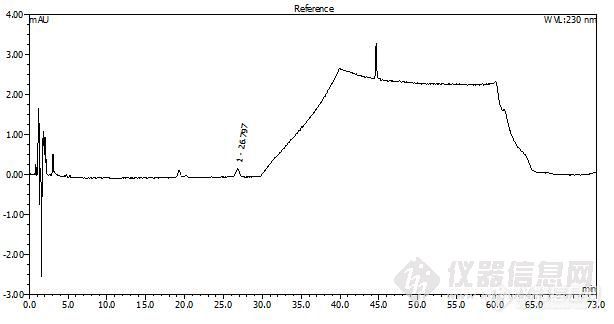
[size=3][font=宋体]各位有做这个项目的么? 按照药典相关规定,摸索梯度洗脱条件得到下图。但是按照药典规定,主成分色谱峰的峰高为满量程的20%(见下文红色标示)。可是在本色谱条件下,梯度系统峰的变化比苯甲酸雌二醇的主峰峰高大多了,如果按照药典规定出图,30min以后就看不到基线了。“【[/font][font=黑体]检查[/font][font=宋体]】[/font][font=Times New Roman] [/font][font=黑体]有关物质[/font][font=Times New Roman] [/font][font=宋体]用内容量移液管精密量取本品适量(约相当于苯甲酸雌二醇[/font][font=Times New Roman]2mg[/font][font=宋体]),置[/font][font=Times New Roman]100ml[/font][font=宋体]量瓶中,加无水乙醇适量,充分振摇,待溶液澄清后,用无水乙醇稀释至刻度,摇匀,作为供试品溶液。精密量取[/font][font=Times New Roman]1ml[/font][font=宋体],置[/font][font=Times New Roman]100ml[/font][font=宋体]量瓶中,用无水乙醇稀释至刻度,摇匀,作为对照溶液。照苯甲酸雌二醇有关物质项下的色谱条件,量取对照溶液[/font][font=Times New Roman]20μl[/font][font=宋体]注入液相色谱仪,调节检测灵敏度,[color=#fe2419]使主成分色谱峰的峰高约为满量程的[/color][/font][color=#fe2419][font=Times New Roman]20%[/font][/color][font=宋体][color=#fe2419]。[/color]再精密量取供试品溶液与对照溶液各[/font][font=Times New Roman]20µ l[/font][font=宋体],分别注入液相色谱仪,记录色谱图。供试品溶液的色谱图中如有杂质峰,单一杂质峰面积不得大于对照溶液主峰面积([/font][font=Times New Roman]1.0%[/font][font=宋体]),各杂质峰面积的和不得大于对照溶液主峰面积的[/font][font=Times New Roman]1.5[/font][font=宋体]倍([/font][font=Times New Roman]1.5%[/font][font=宋体])(供试品溶液中任何小于对照溶液主峰面积[/font][font=Times New Roman]0.01[/font][font=宋体]倍的色谱峰可忽略不计)。”[/font][/size][img=middle]http://ng1.17img.cn/bbsfiles/images/2010/02/201002071025_200720_1632027_3.jpg[/img]
我现在在做17α-雌二醇和17β-雌二醇的降解,经过一定时间的降解,然后将溶液中残留浓度提取出进行HPLC测定,我家的浓度是2mg/kg的雌激素(分为黑土中、红壤中、北京潮土三种基质),过了一个小时后,我在提取,我用的是超声提取,先加入20ml甲醇,提取30min,再加入10ml超声30min 中,后合并抽滤,就是总体积30ml合并后直接抽滤,然后抽滤完后我就去测定了,但是检测不出来,平行的都是如此,检测不出来,是不是因为体积太大,浓度太小,造成的?那我需要旋蒸干后,再加入10ml甲醇,再抽滤监测还是?
我用LC-MS 测定雌二醇、雌三醇、雌酮 ,在摸索质谱条件时 ,总找不到碎片离子 ,ESI+和ESI-都试过 参考文献用别人的质谱条件 ,做液质联用时不出峰 ,别人用的是C18的柱子 ,我们用的是C8 是柱子的问题吗 ?还是质谱的问题 ?我应该怎么解决呢
初步调研市售纯牛奶雌二醇(E2)和孕酮(P4)含量现状。方法:收集同季节6 个品牌(A、B、C、D、E 和F 品牌,各品牌随机采集6 个批号)共36 个批号市售纯牛奶,采用免疫化学发光分析法检测经硫酸酯酶水解的乳清总E2(TE2)和P4。结果:6 个品牌乳清TE2 差异无统计学意义;F 品牌乳清P4 高于C 品牌(P < 0.05),其余各品牌间差异均无统计学意义。36 个批号中,乳清TE2 和P4 最大值与最小值之比分别为2.32 和6.67。A、B、C、D、E 和F 品牌批号间乳清TE2 的最大值与最小值之比分别为1.38、1.60、1.82、1.79、2.17 和1.68,其P4的最大值与最小值之比分别为3.19、5.98、2.89、1.38、1.86 和1.67。6 个品牌的乳清TE2 和P4 均值的最大值与最小值之比分别为1.25 和3.05。结论:市售纯牛奶品牌间乳清P4 存在差异,且同品牌不同批号波动较大;批号间乳清TE2 也有一定差异。
各位专家,大家好,我现在用GC112A分析乙酸丁酯和2,3-丁二醇,柱子用的是SE-30和SE-54,2,3-丁二醇沸点为181左右,乙酸丁酯为126.1,可是乙酸丁酯的峰在2,3-丁二醇后面,而且分离度不够,不能大于1.5,我采用了各种升温程序都不行,有谁能告诉我怎么办么??
我现有一产品需要做有机溶剂残留量,可能的残留有机溶剂主要为二氯甲烷,乙酸乙酯,甲醇,乙醇.请问应该用什么样的色谱条件?
介绍一篇用NMR和X-RAY测定17-beta-雌二醇结构的结果比较的文章,同样一个化合物,用NMR(1D和2DNMR)和X-RAY测定时得到一些不同的结构信息(当然化合物结构没问题的),就是说,一个化合物在固体和/或结晶状态与溶液状态的分子"形状"是有区别的,主要反映在构象上,在研究生物活性时,溶液状态更接近生物提内的情况.有兴趣者可以参考,讨论.17-beta-雌二醇结是临床上的一种甾体药物.[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=4460]相关附件[/url]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=125076]丙二醇单甲醚乙酸酯的合成[/url]
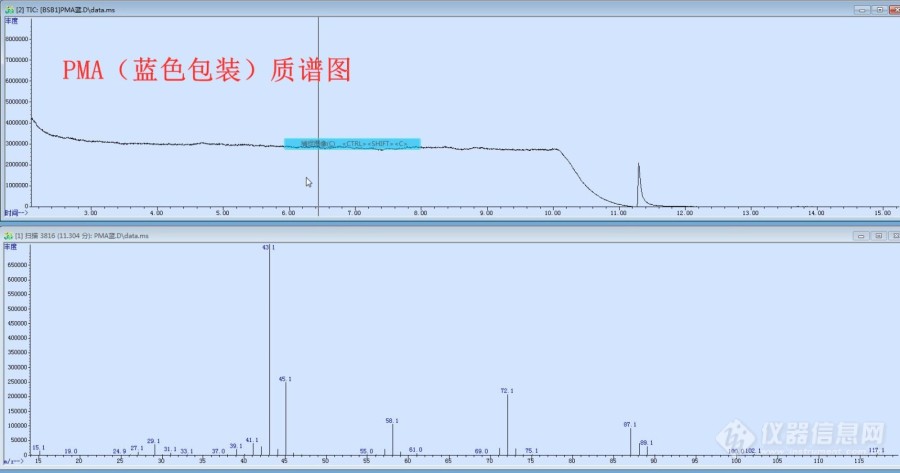
2018年,无意中发现仓库里的两个批次PMA出峰时间有一点区别(一批次是白色桶包装,另一批次是蓝色桶包装,是同一个厂家生产的),当时闻了两个批次PMA的气味是一样的,就没有在意,只是把两个批次的PMA分别留了一瓶样, 前几天把2018年留的样拿出来重新测了一下, 一、色谱上检测,两个样品确实出峰时间不一样,(下图色谱图是两个样1:1混合,出两个峰,峰面积差不多1:1)。 二、质谱走样, 1、正常PMA(蓝色桶)主要离子丰度从大到小大致是43、45、72、58、87,与谱库离子对的上号。 2、未知物(白色桶PMA)主要离子丰度从大到小大致是59、43、29、31、72。 三、百度上搜索了下,丙二醇甲醚乙酸酯好像没有其他结构,只有在HG/T3940-2007工业用丙二醇甲醚乙酸酯标准中提到过“2-甲氧基-1-丙醇乙酸酯”的物质请专家指导一下,从质谱上分析,这个未知物(白色桶PMA)会不会是2-甲氧基-1-丙醇乙酸酯?[img=,690,473]https://ng1.17img.cn/bbsfiles/images/2021/06/202106011541055349_1792_2932653_3.png!w690x473.jpg[/img][img=,690,362]https://ng1.17img.cn/bbsfiles/images/2021/06/202106011541232724_8127_2932653_3.jpg!w690x362.jpg[/img][img=,690,358]https://ng1.17img.cn/bbsfiles/images/2021/06/202106011541376269_8104_2932653_3.jpg!w690x358.jpg[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2021/06/202106011541500369_5011_2932653_3.jpg!w690x920.jpg[/img]
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
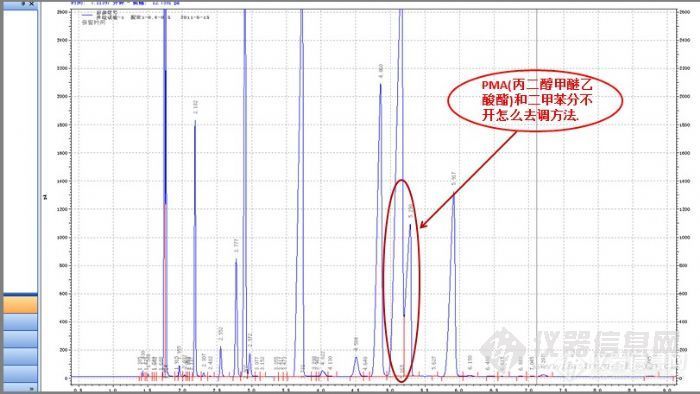
测涂料里的苯系物 PMA+二甲苯分不开各位: 1、我今天做了一下PU白面漆的苯系物含量,但做出来的谱图里面的 PMA(丙二醇甲醚乙酸酯)和那个二甲苯分不开,要怎么去调方法呢? 2、做涂料的苯系物含量是不是要先按要求配比好(漆:固化剂:稀释剂),再加一点乙酸乙脂稀释,再离心一下,取上部清液分析呢。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191653_630872_2263490_3.jpg
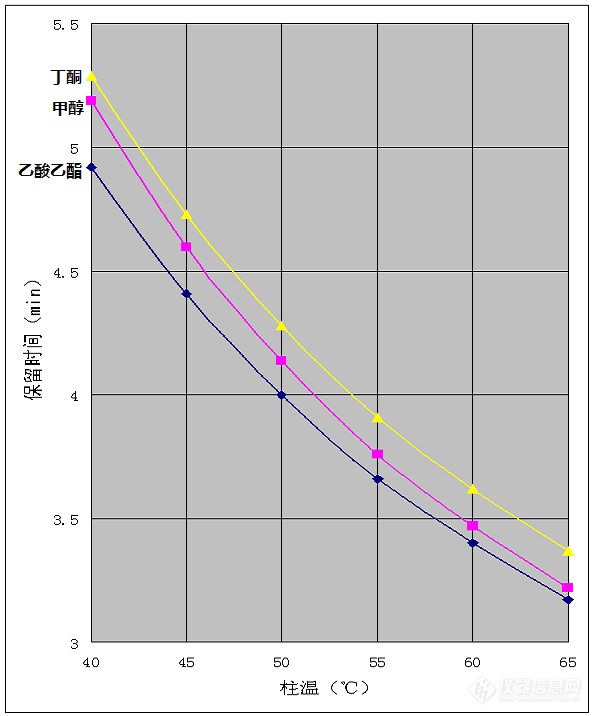
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
题目:注射用载雌二醇m PEG-PLA缓释微球的制备及体内外释放研究作者:李静期刊:山东大学,2012链接:http://www.cnki.net/KCMS/detail/detail.aspx?dbcode=CMFD&QueryID=0&CurRec=1&dbname=CMFDTEMP&filename=1012462466.nh&urlid=&yx=
se54毛细柱可用于检测丙二醇甲醚乙酸酯吗
se54毛细柱子可用于检测丙二醇甲醚乙酸酯吗
请问DMBC Acetate乙酸二甲基苄基原醇酯的质谱图是怎样的?CAS000151-05-3
产物中有空气,二氧化碳,乙醇,乙醛,乙酸,乙酸乙酯,之前一直用PQ柱,乙酸响应太低,且拖尾严重,求推荐合适的填充柱。
用于氯霉素、克伦特罗和雌二醇三种兽药残留检测的高通量悬浮芯片技术研究摘要:目的建立一种氯霉素、克伦特罗和雌二醇(17-beta-estradiol,E2)的3种兽药残留的新型高通量悬浮芯片检测技术。方法:合成3种兽药的牛血清白蛋白(bovine serum albumin,BSA)结合物,并进行紫外和质谱鉴定。绘制出3种兽药残留检测的标准曲线;对不同浓度的干扰物和待测物分组,以此进行特异性检测和盲样测定。并用扫描电子显微镜(简称电镜)进行微球表面微观结构观察。悬浮芯片检测的标准曲线方程和方程相应的决定系数(R^2)表现良好,R^2〉0.99;3种兽药悬浮芯片的检测区间分别为(40.00~6.25)×10^5ng/L,(50.00-7.81)×10^5ng/L和1.00×10^3~7.29×10^5ng/L;最低检出限为:40ng/L、50ng/L和1μg/L;同时,悬浮芯片的特异度测试良好,与其他药物无明显交叉反应;对盲样测定的检测浓度值与实际浓度偏差在8.09%-17.03%,可认为偏差较小。原文:资料中心。
请问乙二胺四乙酸分析纯能直接配制EDTA标准溶液吗?请如果能配制有方法吗,比如配0.1mol/L的,请各位帮忙,我手里现在只有乙二胺四乙酸分析纯
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱做邻苯二甲酸酯类(二丁酯、二乙基己酯),进溶剂空白(甲醇、乙腈、二氯甲烷、乙酸乙酯,均为色谱纯或农残级),空白均有很高响应,以致标准曲线都无法做,选择离子扫描,149出很大峰,希望各位专家能有解决办法,谢谢!急!
在对黄连进行薄层色谱的时候,用环己烷:乙酸乙酯:异丙醇:甲醇:水:三乙胺(3:3.5;1:1.5:0.5:1)进行展开,但是没有展开完,就感觉有二次展开的情况,想问问出现这种情况的原因,和解决方法
我今天按照国标GB/T5750.10-2006中的9测定二氯乙酸,进行的是加标回收。因为是用甲基叔丁基醚做的介质,质谱分析时有很多峰,我无法确定哪一个是二氯乙酸甲酯,自带的质谱库内也没有。请问哪位能帮我找一下二氯乙酸甲酯的质谱分析参数,谢谢。
二硫化碳毒性太大了,实验室也没准备,是上气相色谱法做乙酸乙酯、己酸乙酯用的,可以用乙醇稀释吗?有人做过吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP