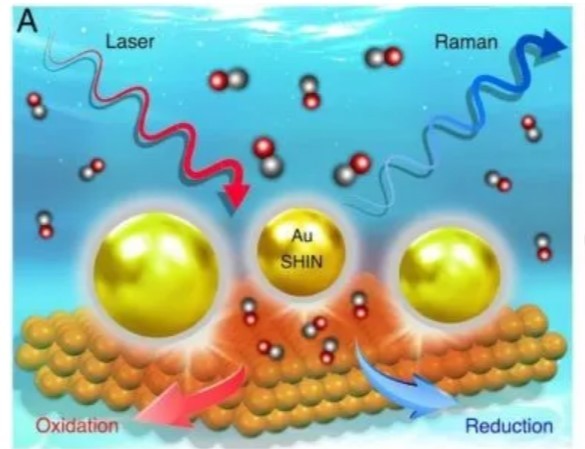
东京大学化学家首次原子分辨透射电镜制作化学合成分子视频
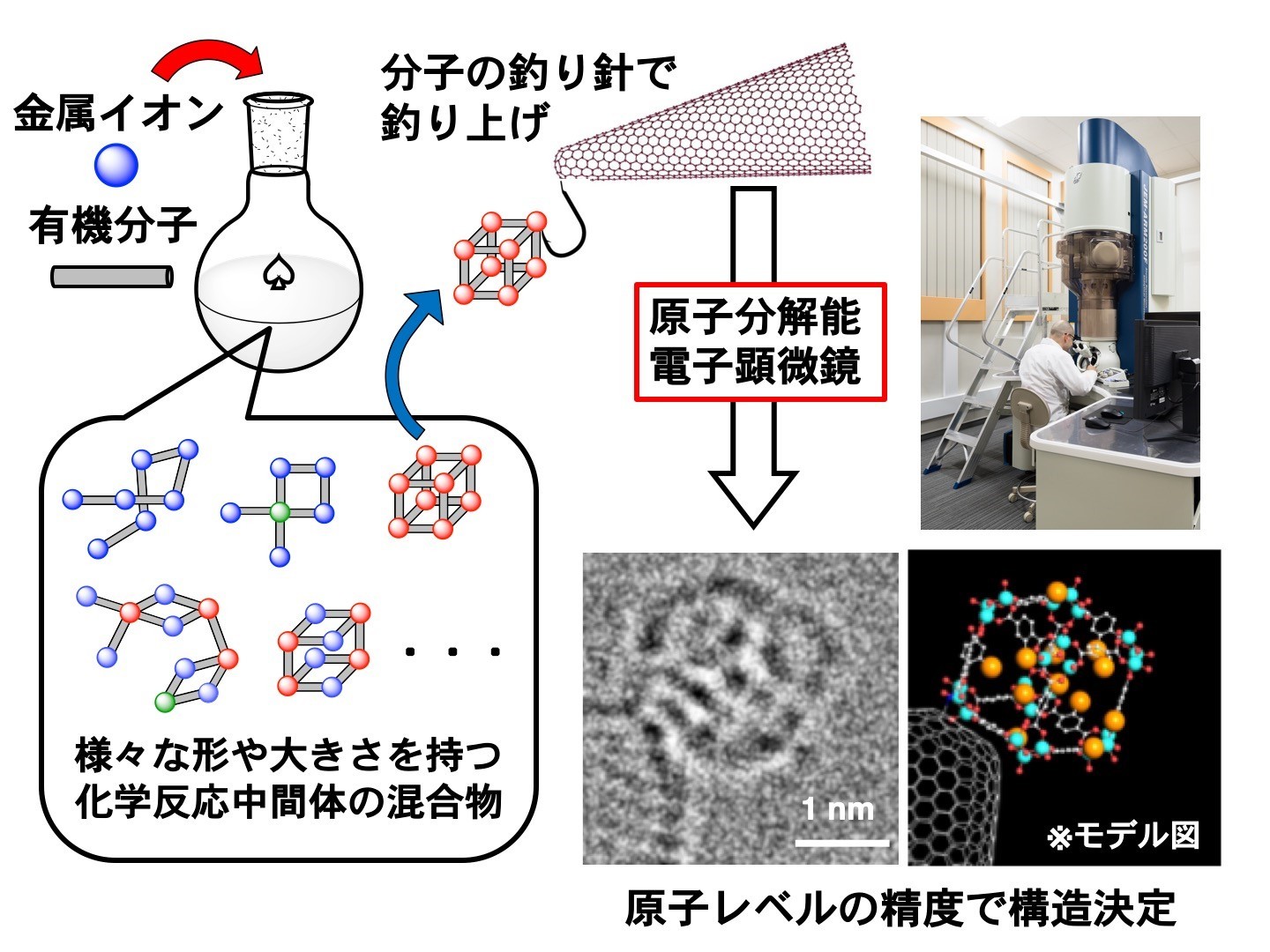
p strong 仪器信息网讯 /strong 8月23日,东京大学中村荣一(化学系特聘教授/东京大学名誉教授)、原野幸治(化学系特任副教授)合作在Nature Communications刊发文章《Atomistic structures and dynamics of prenucleation clusters in MOF-2 and MOF-5 syntheses》( /p p br/ /p p DOI:10.1038/s41467-019-11564-4 ),首次以原子分辨率透射电镜制作出化学合成视频。 /p p span style=" background-color: rgb(255, 0, 0) color: rgb(255, 255, 255) " strong 发表者点评 /strong /span /p p strong 中村荣一 /strong 教授表示:“自2007年以来,物理学家已经实现了超过200年的梦想, 能够看到单个原子的能力。但这并没有就此结束。我们的研究小组已经超越了这个梦想,创造了分子视频,以前所未有的细节观察化学反应。” /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 450px height: 338px " src=" https://img1.17img.cn/17img/images/201908/uepic/72b8ccad-2767-4e1a-8cdd-e5abaf43c6a6.jpg" title=" 00.jpeg" alt=" 00.jpeg" width=" 450" height=" 338" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 东京大学化学系教授中村荣一 /span /p p strong 原野幸治 /strong 解释道:“这是一个两部分的问题。在宏观上,将独特的高分辨率电子显微镜与快速灵敏的成像传感器结合起来进行连续视频成像存在工程挑战 而在微观层面上,我们必须设计一种方法来捕获感兴趣的分子,把它们固定到位,这样相机就能捕捉起运动作用。” /p p “让我们感到惊讶的是我们的计划确实有效。这是一项复杂的挑战,但我们首先在2013年对这些分子视频进行了视觉化。从那时到现在,我们努力将这个概念变成一个有用的工具。我们的首个成功是成像和描述一个立方体形状的分子,这是金属-有机骨架合成过程中发生的一种重要的中间形式。我们用了一年时间来说服我们的论文审稿人我们发现的是真实的。” /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 450px height: 270px " src=" https://img1.17img.cn/17img/images/201908/uepic/13ff28e1-e633-48ec-a361-42e4f3160e29.jpg" title=" 0.jpeg" alt=" 0.jpeg" width=" 450" height=" 270" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 原野幸治与研究中使用的原子分辨率透射电子显微镜 /span /p p strong span style=" background-color: rgb(255, 0, 0) color: rgb(255, 255, 255) " 发表要点 /span /strong /p p 在溶液中捕捉化学反应中一个接一个地产生和消失的中间产物(反应中间体)的每一个分子,并且用电子显微镜观察到并确定了迄今为止未知的反应中间体的结构。 /p p 用以往的分析方法,只能对溶液中发生的各种化学反应中间体的混合物的平均分子图像,或极少一部分的分子图像进行分析。本次提取了每一个分子,并成功确定了结构。 /p p 此次研究表明,此研究方法可以确定以往常规方法无法观察到的化学反应中间体的每一个分子的结构,从材料科学到生物化学有望得到广泛的学术和工业应用。 /p p span style=" color: rgb(255, 255, 255) background-color: rgb(255, 0, 0) " strong 发表概要 /strong /span /p p 东京大学化学系教授中村荣一和副教授原野幸治等研究小组开发了中间产物(反应中间体),它们在化学反应中一个接一个地产生和消失。在溶液中捕获并且通过原子分辨率电子显微镜(电子显微镜,注释1)观察,成功地确定了迄今未知的反应中间体的结构。 /p p ( span style=" color: rgb(165, 165, 165) " 注释1:像差校正技术的最新进展使得即使使用适于观察有机材料的低加速电压的电子显微镜也能够以原子分辨率捕获图像。 2015年在东京大学分子生命创新大楼新建立的最先进的透射电子显微镜实现了超高速连续拍摄,空间分辨率为0.08纳米,每秒1600幅图像。 /span ) /p p 化学反应一般在从反应物到生成物的过程中,通过不断形成系列反应中间体推动进行。这些中间体的结构各不相同,而且由于在反应溶液中保持平衡,结构不断变化,所以很难通过实验捕获结构。中村教授等,将对反应中间体具有很强亲和力的“分子鱼钩”装在碳纳米管上,再将纳米管放入反应溶液中进行反应。然后,开发了一种新技术,通过快速冷却和过滤快速停止反应,将在反应进行的各阶段发生的一系列的中间体“抓住在鱼钩”上,一网打尽地依次进行结构分析(图1)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 450px height: 338px " src=" https://img1.17img.cn/17img/images/201908/uepic/f4f19cfd-7e1b-4012-b0a0-6a10f0e90416.jpg" title=" 1.jpg" alt=" 1.jpg" width=" 450" height=" 338" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图1.本研究中使用的“分子钩”技术的概念图 /span /p p 本次研究中采用的反应是气体储存材料和催化剂形成金属-有机骨架(MOF)的反应,反应中间具有一维至三维结构(簇),并且从原子水平,统计信息阐明了微小分子聚集体生长成晶体的反应机理。 /p p 除人工化学合成反应外,本方法还可应用于对天然矿物、骨矿等矿物质生成等材料形成的反应分析,有望开发出高效化学反应、阐明生命现象。 /p p strong span style=" background-color: rgb(255, 0, 0) color: rgb(255, 255, 255) " 发表的内容 /span /strong /p p strong 研究背景 /strong /p p “像观看分子模型一样观察分子的反应情况”是科学家长期以来的梦想,也是极其困难的课题。在化学反应中,在将一种物质转换为另一种物质的过程中,存在大量的中间生成物(反应中间体),并且作为混合物形式存在。 /p p 在现有化学反应的分析中,反应总体情况的一般的描述方法是:这些众多反应中间体的平均值,或者主要反应中间体被分离后的结构分析。而每个中间体有各自的形状和大小,关于它们的每种形状和大小的信息并无从获得。特别是,当许多物质参与化学反应时,分析更加困难。为了阐明化学反应机理的细节,有必要建立一种澄清由分子之间的微小反应产生的每个纳米级中间体的结构的方法。 /p p 中村教授等人的研究小组在2007年以来,“它单分子原子分辨率实时间电子显微镜(smart-em)映射”的独立开发的分子运用电子显微镜技术,小分子一个一个的动态视频拍摄记录的研究正在进行。2012年,有机分子的结晶化过程中产生的分子集合体的分子结构及出现频率决定成功,视频拍摄,但单分子不仅分子集合体的研究中也史无前例的最尖端的测量法和报告(nat . mater . 2012, 11, 877)。 /p p 自2007年以来,中村教授等的研究小组充分利用了独立开发的分子电子显微镜技术“原子分辨率单分子实时电子显微镜(SMART-EM)成像”(注4),“小分子一个一个的动态视频拍摄记录的研究正在进行”。2012年,研究小组成功地拍摄确定了有机分子结晶过程中分子组装的分子结构和出现频率,视频拍摄在分子组装和单分子研究中前所未有。相关报道称这是一种最尖端的测量方法(Nat.Mater.2012,11,877)。 /p p strong 具体的研究内容 /strong /p p 这次,中村教授研究小组, 在碳纳米管尖端引入了化学亲和力,并用它作为“分子钩”从反应混合物中拾取反应中间体,然后使用原子分辨率进行结构解析。通过显微观察成功地以惊人的方式分析了结构(图1)。此外,表明可以基于所获得的数百种反应中间体的结构的统计信息来研究反应机理。 /p p 在这项研究中,主要专注于一组称为金属有机框架(MOF)的物质。MOF内部具有规则的纳米孔,其在储气剂和催化剂中的应用已得到广泛研究,但MOF形成过程的实验信息极为有限。尤其是MOF形成初期发生的纳米尺寸的反应中间体的结构信息,目前尚未获得。因此,这次,由对苯二甲酸(PET瓶的原料)和硝酸锌(图2)合成的两种MOF(称为MOF-2和MOF-5)用于制备具有对苯二甲酸作为分子钓鱼钩的碳纳米管。通过与尖端结合来拾取反应中间体,并进行结构分析(图3)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 450px height: 338px " src=" https://img1.17img.cn/17img/images/201908/uepic/80fb40ca-4192-4136-a008-9153d11fc727.jpg" title=" harano_2.jpg" alt=" harano_2.jpg" width=" 450" height=" 338" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图2.通过在溶液中加热对苯二甲酸和六水合硝酸锌而生产的两种MOF(MOF-2和MOF-5)。 MOF-2具有通过溶剂层叠网状平面网络的结构,而MOF-5具有像丛林健身房那样的三维网状结构,并且两种结构都具有纳米尺寸的孔。 下部显示晶体结构。 浅蓝色,红色和灰色球体分别代表锌,氧和碳原子。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 450px height: 247px " src=" https://img1.17img.cn/17img/images/201908/uepic/fd333a83-18cf-45e3-9623-ba98d8c16f4e.jpg" title=" harano_3.jpg" alt=" harano_3.jpg" width=" 450" height=" 247" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图3.使用附着在碳纳米管尖端上的“分子钓鱼钩”来升高MOF形成反应中间体的反应示意图。 /span /p p 在反应的每个阶段通过在每个MOF产生、反应、猝灭然后停止反应的温度条件下将碳纳米管与分子钩一起添加到反应混合物中而产生的一系列中间体被鱼钩抓住。然后,将提取的碳纳米管置于电子显微镜中真空条件下观察,以原子分辨率获得MOF形成的反应中间体而产生的1-2纳米尺寸的聚集体(簇)。松散耦合的簇在电子显微镜拍摄的实时尺度上是自发旋转的,因此,可以不倾斜样品而从各种角度观察其三维结构,并在原子水平上揭示了其三维结构。在MOF的形成过程中,产生了由锌离子和对苯二甲酸构成的无数中间体,但是通过取出一个这样的未知分子来确定结构,对于分子结构分析具有重要意义。 /p p 作为MOF-2的反应中间体,除了许多一维链簇之外,还观察到具有作为MOF-2的部分结构的正方形结构的簇(图4)。另一方面,从MOF-5反应溶液中拾取具有立方三维结构的中间体(图5)。通过使用含有碘的对苯二甲酸作为重元素,可以使用碘作为标记物来确定三维结构,并且含有12个有机分子的立方中间体小于1埃,可以精度确定结构(图6)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 230px " src=" https://img1.17img.cn/17img/images/201908/uepic/3f5be26a-4141-464f-9cd4-9e741caaf74b.jpg" title=" harano_4.jpg" alt=" harano_4.jpg" width=" 600" height=" 230" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图4.从MOF-2反应溶液收集的碳纳米管尖端拾取的反应中间体的电子显微镜图像。 (左)许多一维链簇(箭头)和二维方阵(包围虚线)。 (右)从触摸钟摆的方形簇的电子显微镜电影中提取的每个帧。顶部是真实图像,底部是相应的分子模型。图中的数字是从视频录制开始经过的时间(单位:秒)。原子着色与图1中的相同。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 241px " src=" https://img1.17img.cn/17img/images/201908/uepic/5a962d8f-8b0c-4018-8eaa-b8c9dfb958cd.jpg" title=" harano_5.jpg" alt=" harano_5.jpg" width=" 600" height=" 241" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图5.用碘原子标记的MOF-5立方反应中间体的原子分辨率电子显微镜电影。图中的数字是从视频录制开始经过的时间(单位:秒)。图中的比例尺为1纳米。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 424px " src=" https://img1.17img.cn/17img/images/201908/uepic/2bb411ce-c5d7-4a49-a86d-5a1a4caaebac.jpg" title=" harano_6.jpg" alt=" harano_6.jpg" width=" 600" height=" 424" border=" 0" vspace=" 0" / /p p style=" text-align: center " span style=" color: rgb(0, 176, 240) " 图6示出了从图5所示的运动图像中提取的电子显微镜图像的帧(左),与每个图像对应的分子模型(右),以及电子显微镜模拟图像(中心)。图中的数字是从视频录制开始经过的时间(单位:秒)。图中的比例尺为1纳米。 /span /p p 这只是能够以精确和可控的方式控制化学合成的第一步,研究人员称之为“示构合成”。随着合成反应的进步,观察反应的细节非常重要,这样才能有效地进行逆向工程。 /p p 化学家200年前的梦想是看到一个原子,现在的梦想是控制分子,以便建筑创造出合成矿物质,甚至是拯救生命的新药。 /p p strong 附: /strong /p p 本研究的电子显微镜的部分图像分析是在日本科学技术厅(JST)研究成果展开事业尖端测量技术和设备开发计划(课题编号:JPMJSN16B1)的支援下实施的。 /p p 在这项研究中,使用原子分辨率透射电子显微镜(JEM-ARM200F,由JEOL Ltd.制造),这是由国际科学创新中心开发项目引入并由东京大学分子生命创新组织运营的共享仪器。透射电子显微镜观察的一部分是在教育,文化,体育,科学和技术部纳米技术平台的支持下进行的(发行编号:12024046) /p p strong 论文链接: /strong a href=" https://www.nature.com/articles/s41467-019-11564-4" target=" _blank" style=" color: rgb(0, 176, 240) text-decoration: underline " span style=" color: rgb(0, 176, 240) " https://www.nature.com/articles/s41467-019-11564-4 /span /a /p























 我要推广仪器
我要推广仪器
 下载APP
下载APP