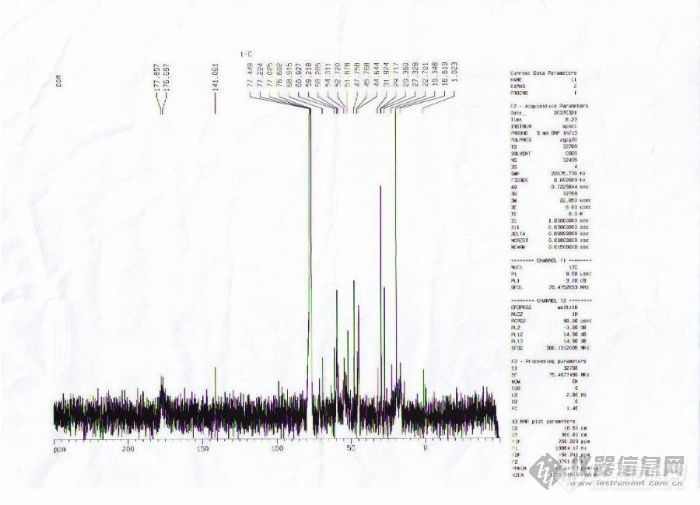
是4-5个甲基丙烯酸酯类共聚物的,两张不同地方做的谱图,求高手帮助分析,也可以通过站内短信联系。[img]http://ng1.17img.cn/bbsfiles/images/2008/12/200812211722_125489_1608602_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2008/12/200812211722_125490_1608602_3.jpg[/img]
表征共聚物的分子量及分子量分布通常要用到GPC,但是我是做接枝共聚物的,聚合物的结构与PS标样相差很大。因此导致做出来的数据很难说清楚其物理意义。因此恳请大家来谈一谈GPC表征接枝共聚物的优劣,改进方法以及替代方法等。热切期待中.......................
预聚物、调聚物、齐聚物、缩聚物、共聚物、均聚物的概念 1、预聚物聚合度介于单体与最终聚合物之间的一种分子量较低的聚合物,通常指制备最终聚合物前一阶段的聚合物。2、调聚物在聚合反应中,如ktr(链转移速率常数) kp(再引发速率常数),则形成聚合度很小的低聚物,这类反应称做调聚反应,因此这种调聚反应得到的聚合物也称为调聚物。其分子量较低,一般只有二到十个链节,分子的两端是与调聚剂分子分裂部分结合的。如果新自由基活性减弱,则再引发相应减慢,会出现缓聚现象,聚合速率和聚合度都将显著降低。极端的情况是新自由基稳定,难以继续再引发增长,就成为阻聚作用。3、齐聚物又称低聚物。高分子与低分子的区别在于前者分子量很高,通常将分子量高于约1万的称为高分子(polymer),分子量低于约1000的称为低分子。分子量介于高分子和低分子之间的称为低聚物(oligomer,又称齐聚物)。一般高聚物的分子量为104~106,分子量大于这个范围的又称为超高分子量聚合物。但是在行业中,比如PAM,分子量在1500~1800万以上的才称为超高分子量PAM。4、缩聚物生成聚合物时有水或其他简单分子放出的聚合称为缩聚,用这种方法合成的聚合物称为缩聚物。5、共聚物两种或两种以上的单体或单体与聚合物间进行的聚合称为共聚,共聚得到的产物即为共聚物。分嵌段共聚物、接枝共聚物、无规共聚物、有规共聚物等。6、均聚物由一种单体聚合而成的聚合物称为均聚物。P.S:英文的“高分子”主要有两个词,即polymer和macromolecule。前者又可译作聚合物或高聚物;后者又可译作大分子。这两个词虽然常混用,但仍有一定区别,前者通常是指有一定重复单元的合成产物,一般不包括天然高分子,而后者指分子量很大的一类化合物,包括天然和合成高分子,也包括无一定重复单元的复杂大分子。
1、预聚物聚合度介于单体与最终聚合物之间的一种分子量较低的聚合物,通常指制备最终聚合物前一阶段的聚合物。2、调聚物在聚合反应中,如ktr(链转移速率常数) kp(再引发速率常数),则形成聚合度很小的低聚物,这类反应称做调聚反应,因此这种调聚反应得到的聚合物也称为调聚物。其分子量较低,一般只有二到十个链节,分子的两端是与调聚剂分子分裂部分结合的。如果新自由基活性减弱,则再引发相应减慢,会出现缓聚现象,聚合速率和聚合度都将显著降低。极端的情况是新自由基稳定,难以继续再引发增长,就成为阻聚作用。3、齐聚物又称低聚物。高分子与低分子的区别在于前者分子量很高,通常将分子量高于约1万的称为高分子(polymer),分子量低于约1000的称为低分子。分子量介于高分子和低分子之间的称为低聚物 (oligomer,又称齐聚物)。一般高聚物的分子量为104~106,分子量大于这个范围的又称为超高分子量聚合物。但是在行业中,比如PAM,分子量在1500~1800万以上的才称为超高分子量PAM。4、缩聚物生成聚合物时有水或其他简单分子放出的聚合称为缩聚,用这种方法合成的聚合物称为缩聚物。5、共聚物两种或两种以上的单体或单体与聚合物间进行的聚合称为共聚,共聚得到的产物即为共聚物。分嵌段共聚物、接枝共聚物、无规共聚物、有规共聚物等。6、均聚物由一种单体聚合而成的聚合物称为均聚物。P.S:英文的 “高分子”主要有两个词,即polymer和macromolecule。前者又可译作聚合物或高聚物;后者又可译作大分子。这两个词虽然常混用,但仍有一定区别,前者通常是指有一定重复单元的合成产物,一般不包括天然高分子,而后者指分子量很大的一类化合物,包括天然和合成高分子,也包括无一定重复单元的复杂大分子。
[em0812] 拜托大家,有谁知道丙烯共聚物的一些表征方法(除了IR、DSC、NMR、TREF、SM、GPC、X-ray外),这些在我们实验室做不了,能不能有一些简单点的,例如用正庚烷萃取测等规度,二甲苯可溶物的测定。
大家好: 我现在有个离子液体样品,里面含有大概1%的N-甲基咪唑量,因为用气相色谱做这个样品对柱子有损坏作用,现在想通过液相色谱法对N甲基咪唑做定量分析。我公司有安捷伦的液相色谱,柱子是C18的,检测器有紫外和视差检测器。我们自己通过液相实验后分析不出样品中的N甲基咪唑,但进纯样N甲基咪唑后有信号峰出现。想求教下大家有什么好方法能分析出离子液体样品中的N甲基咪唑。
有网友问:做4-甲基咪唑不出峰。如何解决。
是几个Tg还是说几个熔点,或其他的。如何说明我合成的聚合物就是嵌段成功了呢?主要是用于区别于无规共聚物或共混物
请问嵌段共聚物用核磁如何表征?
甲醛苯胺共聚物中的苯胺和甲醛在红外光谱上油特征峰吗?能否进行定量?
reach中的甲醛苯胺共聚物如何进行测试?
接枝共聚物自组装成球状结构,可通过核磁来确认核和壳的结构吗?如何操作可以得到比较可靠信息呢?
[em0706] 请教熟手:我是新手,想学习凝胶色谱,请指点。1目前单位分析的对象是一系列丙烯酸共聚物(分子量6000左右),制作样品的方案是:第一步:0.1克左右,用丁酮溶解,电热板烘干(不明白这一步的意义);第二步,用流动相(四氢呋喃)按100倍稀释,然后进样60UL.请说明,谢谢。1如果测试笨乙烯试样应该怎样配置样品。谢谢。(窄分布标样是苯乙烯)
共聚时应该会有新的吸收峰出现,那么原有的吸收峰的位置是否会发生移动?比如丙烯酰胺和丙烯酸共聚物的红外光谱中属于丙烯酰胺单元的吸收峰和丙烯酰胺均聚物的红外光谱中的相应吸收峰位置是否重合? 如果不重合,那么是否可以通过吸收峰位置的改变说明原有的聚合物链上出现了别的基团? 不好意思!我是个外行,问的问题可能有点幼稚,希望大家多多指点。
请教大家我用国标方法做4-甲基咪唑,回收率只有20%是什么原因?我的过程是:取15mL5mg/L的4-甲基咪唑标准品,加1mol/L的碳酸钠5mL, 混匀,加入20mL80:20的三氯甲烷和乙醇混合物,摇荡提取,弃去水层,再用20mL50mM的甲烷磺酸液提取,过滤上机,可是为什么回收率这么差呢?
如何用DSC区别乙丙共聚物与聚乙烯/聚丙烯共混物
[align=center][font='times new roman'][size=16px]苯乙烯[/size][/font][font='times new roman'][size=16px]-[/size][/font][font='times new roman'][size=16px]马来酸共聚物[/size][/font][font='times new roman'][size=16px]及其应用[/size][/font][/align] 苯乙烯与马来酸酐的[back=#ffffff]共聚物[/back][back=#ffffff]苯乙烯[/back][back=#ffffff]-[/back][back=#ffffff]马来酸([/back][back=#ffffff]SMA[/back][back=#ffffff])[/back][back=#ffffff]首先由[/back][back=#ffffff]Alfred[/back][back=#ffffff]和[/back][back=#ffffff]Lavin[/back][back=#ffffff]在[/back][back=#ffffff]1945[/back][back=#ffffff]年制[/back][back=#ffffff]备。[/back][back=#ffffff]之后[/back][back=#ffffff],[/back][back=#ffffff]Mayo[/back][back=#ffffff]等提出[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]共聚体系是典型的交替共聚模型[/back][back=#ffffff],[/back][back=#ffffff]具有强吸电子基团的马来酸酐与具有给电子基团[/back][back=#ffffff]的[/back][back=#ffffff]苯乙烯是一对电荷转移复合物,在自由基引发体系中具有很好的交替共聚特征,但是传统的自由基聚合会导致[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]的聚合不可控且分子量分布较宽等问题,限制了[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]共聚物[/back][back=#ffffff]的应用,“活性”[/back][back=#ffffff]/[/back][back=#ffffff]可控自由基聚合法为[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]的合成提供了解决方案,[/back][back=#ffffff]但是也有着显著区别。[/back][back=#ffffff]对于[/back][back=#ffffff]A[/back][back=#ffffff]TRP[/back][back=#ffffff]法,马来酸酐会与催化剂中金属离子发生反应,导致催化剂失效,因此只能采取光引发等无金属[/back][back=#ffffff]A[/back][back=#ffffff]TRP[/back][back=#ffffff]法合成。对于[/back][back=#ffffff]N[/back][back=#ffffff]MP[/back][back=#ffffff]法,由于聚合所需的温度较高,只能得到[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]的无规[/back][back=#ffffff]则[/back][back=#ffffff]共聚物。利用[/back][back=#ffffff]R[/back][back=#ffffff]AFT[/back][back=#ffffff]法可以较好地进行共聚,并且可以得到交替共聚物。在实际的聚合反应体系中,苯乙烯与马来酸酐的交替共聚速率远大于苯乙烯的自聚速率,并且马来酸酐的自聚能力很低,因此在苯乙烯过量的情况下,会首先形成[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]交替共聚物,此后再是苯乙烯的自聚,最终可形成具有[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]交替和[/back][back=#ffffff]苯乙烯[/back][back=#ffffff]自聚的嵌段共聚物[/back][back=#ffffff]。[/back] [back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]的一个重要优势在于马来酸酐中酸酐基团的高反应活性,可以在较温和的条件下发生酯化、酰胺化等反应,因此可以引入新的功能性基团,得到改性的[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]衍生物,这大大拓展了其应用范围[/back][back=#ffffff]。[/back][back=#ffffff]由于[/back][back=#ffffff]S[/back][back=#ffffff]MA[/back][back=#ffffff]及其衍生物具有独特的两亲性和生物相容性,已经被大量应用于膜蛋白增溶提取、药物递送和新材料合成等领域。[/back] [align=center][font='times new roman'][size=16px]S[/size][/font][font='times new roman'][size=16px]MA[/size][/font][font='times new roman'][size=16px]与膜蛋白质[/size][/font][/align] 在多细胞生物中,膜蛋白约占总蛋白质的三分之一。它们在细胞间信号传导和跨细胞膜转运中发挥着重要作用。2009年Knowles等首次报道了SMA共聚物可以直接将生物膜溶解成脂质纳米圆盘(SMALPs),既保留了圆盘内的蛋白质,又确保了膜蛋白稳定的天然脂质环境。此后,使用SMA共聚物的无去污剂增溶方法被大量应用于从生物膜中直接提取蛋白质和脂质。 目前为止,研究人员发现对于苯乙烯与马来酸组成比为3:1或2:1的共聚物结构对于膜的溶解最有效。以3:1的SMA为例简要描述其增溶机制,首先在阶段1中,苯乙烯单元穿透到磷脂双分子层的疏水部分且马来酸酐与亲水性头基结合,此时SMA从一开始紧凑且聚集的构象转变为解聚、延伸的构象,SMA已经插入到磷脂双分子层中。在阶段2中,SMA在磷脂双层中达到饱和状态,此时SMALPs形成,并与SMA饱和的磷脂双层共存。在第3阶段,SMA饱和的磷脂双层完全转化为SMALPs,磷脂双层全部溶解,SMA分布在磷脂双层中,过量的SMA附着在双层周围,生物膜实现增溶。 [align=center] [/align][align=center][font='times new roman'][size=16px]S[/size][/font][font='times new roman'][size=16px]MA[/size][/font][font='times new roman'][size=16px]衍生物[/size][/font][/align] 随着对SMA增溶机制的深入研究发现,SMA的分子量、化学组成与衍生基团的类型等会影响膜蛋白的提取效率与选择性。此外,由于SMA中马来酸的存在,酸的质子化或者与金属阳离子的络合会导致SMA变得过于疏水而无法维持纳米圆盘的结构,比如Mg[font='times new roman'][sup][size=16px]2[/size][/sup][/font][font='times new roman'][sup][size=16px]+[/size][/sup][/font]的浓度高于10 mM或pH低于6时通常会导致SMA沉淀,从而导致SMALPs分解。为了解决上述问题,研究人员开发了大量SMA衍生物,增加了对于pH与金属阳离子(Cu[font='times new roman'][sup][size=16px]2[/size][/sup][/font][font='times new roman'][sup][size=16px]+[/size][/sup][/font]、Mg[font='times new roman'][sup][size=16px]2[/size][/sup][/font][font='times new roman'][sup][size=16px]+[/size][/sup][/font]、Ca[font='times new roman'][sup][size=16px]2[/size][/sup][/font][font='times new roman'][sup][size=16px]+[/size][/sup][/font])的耐受性,为膜蛋白与膜脂的研究提供了更多的选择。例如,Brady等发现2-丁氧基乙醇功能化的SMA衍生物可以促进膜蛋白从蓝藻类囊体膜的提取,而未功能化的SMA基本上是无效的,且较长的疏水性烷氧基乙氧基化物侧链可以提高增溶效率。Burridge等同时合成了SMA-Glu/AE/Neut/Pos四种衍生物,所有的SMA衍生物都能够与以棕榈酰油酰磷脂酰胆碱制备的脂质体反应,形成不同尺寸的SMALPs,都显示出稳定的物理特性,在较宽pH范围和高达100 mM Mg[font='times new roman'][sup][size=16px]2+[/size][/sup][/font]下也可以发挥作用。Lindhoud等通过2-氨基乙硫醇对SMA的部分衍生化,合成了SMA-SH,其可以溶解生物膜,同时SMA-SH中的巯基基团可以与其它活性基团进行衍生化得到新的功能化SMA衍生物,进而实现膜蛋白的选择性提取与纯化,为SMA的应用提供了新思路。 除了对SMA进行衍生化用于提高对膜蛋白的提取效率与选择性之外,部分研究人员也探索了SMA共聚物本身的性质,比如苯乙烯与马来酸酐的比例、链的长度与化学组成分布等,以提高形成SMALPs的能力与稳定性。例如,Cunningham等报道了一种迭代RAFT聚合法合成了具有窄分子量分布与化学组成分布的SMA共聚物。在深入研究之后发现分子量分布与化学组成是影响膜增溶的两个主要因素,宽分子量分布的SMA共聚物,往往具有较高的链长,影响SMA的活性。事实上,较短链长的SMA更有利于SMALPs的形成,因为长链SMA会导致聚合物自身的缠绕,此外长链会同时参与多个SMALPs的形成,进一步影响增溶效率。 [align=center][font='times new roman'][size=16px]S[/size][/font][font='times new roman'][size=16px]MA[/size][/font][font='times new roman'][size=16px]与膜脂[/size][/font][/align] SMA及其衍生物已经广泛应用于膜蛋白的提取与研究。事实上,SMALPs也是用于研究蛋白质周围局部脂质环境的优良体系,但是相关的报道较膜蛋白要少。 Juarez等[font='times new roman'][sup][size=16px][95][/size][/sup][/font]用SMA从两种菌株(野生型N2和细菌抗性菌株agmo-1)中提取脂质,然后通过薄层色谱法和质谱法进行表征,发现从细菌抗性菌株agmo-1中提取的脂质含有醚连接的(O-烷基链)脂质,与仅含有酯连接的(O-酰基)脂质的野生型N2菌株相反。这与细菌抗性菌株agmo-1中功能性烷基甘油单加氧酶(AGMO)的丧失保持一致。此外,与传统的脂质提取方法(需要有机溶剂的方法)相比,SMA可用于生物活体中脂质的提取而不影响其活性,证明了SMA在脂质组学的研究中具有良好潜力。 Rehan等采用电喷雾离子化质谱(ESI-MS)法分析了由SMA提取的人体平衡核苷转运蛋白-1(hENT1)中的脂质组成,因为hENT1是一种需要脂质膜来维持其结构和功能的蛋白质,其周围脂质双层的组成对其活性和稳定性至关重要。分析结果发现,每个hENT1-SMALPs中含有16个磷脂酰胆碱(PC)和2个磷脂酰乙醇胺(PE)脂质分子。除此之外,研究发现使用SMA比使用洗涤剂溶解的hENT1更加稳定。
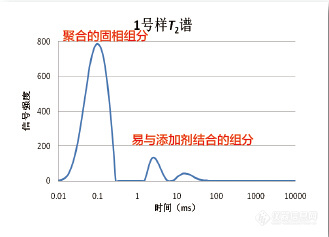
[table=100%][tr][td][align=center][table=100%][tr][td=2,1][align=center][b]小核磁(台式核磁)研究共聚物界面相容性([url=http://www.niumag.com/vtmr.html]相关仪器[/url])[/b][/align][/td][/tr][tr][td=2,1][color=#595757]小核磁(台式核磁)可以提供全面的科研解决方案,适用对象涵盖从橡胶等弹性体 材料到生物领域的膜材料和纳米材料等多种物质。可以利用[b]小核磁(台式核磁)研究共聚物界面相容性[/b]。小核磁(台式核磁)不仅仅提供单个的检测值,无损、快速、便捷的分析过程为工艺改进、过程研究等提供全程、长时间的在线监测。[/color][/td][/tr][tr][td=2,1][align=left][b][color=#595757]以下为用小核磁(台式核磁)研究共聚物界面相容性的部分相关案例[/color][/b][/align][/td][/tr][tr][td=2,1][align=center][img=,329,237]http://ng1.17img.cn/bbsfiles/images/2018/01/201801300917594952_7763_1423_3.jpg!w329x237.jpg[/img][/align][/td][/tr][tr][td=2,1][align=center][b][color=#595757]图一.与添加剂结合后T2弛豫图谱[/color][/b][/align][/td][/tr][tr][td=2,1][align=center][img=,333,236]http://ng1.17img.cn/bbsfiles/images/2018/01/201801300918101302_9239_1423_3.jpg!w333x236.jpg[/img][/align][/td][/tr][tr][td=2,1][align=center][b][color=#595757]图二.与不同添加剂结合后样品的T22弛豫时间[/color][/b][/align][/td][/tr][tr][td=2,1][align=left][b][color=#595757]在高分子材料领域,小核磁(台式核磁)可为您提供以下科研方案[/color][/b][/align][/td][/tr][tr][td=2,1][color=#595757] 1)评价交联聚合体(尤其是橡胶,橡胶产品)的交联信息; 2)评价交联的聚合体(尤其是橡胶,橡胶产品)的物性信息 3)使用过的聚合体材料老化过程的品质鉴定; 4)基于橡胶的硫化,处理和生产条件优化的研究; 5)固体,半硬的聚合体,凝胶体,乳状液和液体的分子活动性研究 6)固体基质中水分和水含量的成像和测定(例如:环氧树脂和半导体器材 7)环氧树脂和橡胶的硫化过程中硫化状态、粘度和过程的探测 8)样品中水或溶液粘合性和活动性的研究 9)聚合物中增塑剂或橡胶含量的测定 10)共混物或共聚物中橡胶含量测定 11)共聚物相对含量测定 12)橡胶胶乳中的固体含量测定 13)临界水及水合作用的研究 14)流变学的的研究,如粘性、密度、及材料的稳定性[/color][/td][/tr][tr][td=2,1][align=left][color=#2778be]使用仪器:[url=http://www.niumag.com/vtmr.html]VTMR20-010V-I核磁共振交联密度成像分析仪[/url][/color][/align][/td][/tr][tr][td=2,1][color=#595757]纽迈专注于“低场核磁共振”技术及应用推广、具备强大的研发能力、完备的生产、服务和成熟的运营管理体系。公司自主开发多款核磁共振分析仪器并已获得多项国家奖项和资质认证,产品广泛应用于农业食品、能源勘探、高分子材料、纺织工业、生命科学等行业领域,获得业界一致认可。[/color][/td][/tr][/table][/align][/td][/tr][/table]
【序号】:1【作者】:张强1李彬2李晓光【题名】:甲基丙烯酸羟乙酯共聚物液体栓塞剂栓塞治疗兔VX2肝肿瘤【期刊】:中国介入影像与治疗学. 【年、卷、期、起止页码】:2018,15(07)【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=1ya23wS0yuB_33nEW3hmN_7djpouki5y9pDaRM8niWu5ote1J4agP-y9o8l-P-d8pYJDwIbn50iWjKwBiGqN03N2c6gbkaTipNZjYdfNd0eMGj3oWHq_wpgH2uZRNvVkdQcEYgx3TDQQG3ye1texXA==&uniplatform=NZKPT&language=CHS

2-甲基咪唑: 分子式 CH3C3H3N2 , 分子量 82.1048, CAS号 693-98-1 性质 固体。熔点142~143℃。沸点267~268℃。溶于水、乙醇、微溶于冷苯。分子式如下:http://ng1.17img.cn/bbsfiles/images/2012/01/201201091849_344855_2357013_3.jpg问题:大家有没有用气相色谱仪测试2-甲基咪唑的方法,或者给出一些相关建议也行,集思广益,希望大家多多发表间接。我自己的做法如下,样品:分析纯2-甲基咪唑0.07g溶于20ml纯水,用FID测试,进样口温度270、柱箱200、检测器270,测试的结果不理想,图谱如下:第一个峰单独进空气验证排除空气峰的可能性,可能是2-甲基咪唑中的杂质峰;第二个峰应该是进样针中残留的2-甲基咪唑,按照此图谱是不是说明可以确认2-甲基咪唑的含量?有版友说此结果只能说明是GC测试的含量,因为样品可能含有一些胺盐FID上是无法响应的,另外2-甲基咪唑含有叔胺和仲胺,这个在FID上的响应是不是也会低一些?http://ng1.17img.cn/bbsfiles/images/2012/01/201201091902_344856_2357013_3.jpg
今年3月,CSPI发布报告称,**可乐等4种产品中,均发现了一种名为4-甲基咪唑的化学物质,应当用何种检测方法?
焦糖色中4-甲基咪唑的测定是采用薄层色谱法,可是最近我在测定样品时,根据标准方法进行操作,却没有标准样品点却没有显色。不知道哪位同仁做过这个检测,烦请帮忙!
我作了一个小实验。利用一种共聚物经过挤出成棒材,植入小耗子的皮下,想观察一下材料的降解情况。在几个固定点取出后测试了DSC,结果发现,熔点逐渐增加,玻璃化温度在逐渐下降。这种情况一般如何解释?通常是哪些原因造成的?在最后一个取出点的时候,其熔点比原材料的熔点还要高(高1℃左右),当然也可以认为是与原材料是一样的。
有机硅/聚氨酯共聚物的制备研究与应用【作者中文名】 窦建芝 陈煜 【文献出处】 化工新型材料 2008年 03期

前不久安谱技术接到一个售前,客户想做4-甲基咪唑,用了HP-5 的柱子 分离情况谱图如下:http://ng1.17img.cn/bbsfiles/images/2014/03/201403301052_494672_1733989_3.jpg用HP-5分离情况不好,安谱的工程师给客户选择了CD-BASEWAX 谱图情况如下,出峰很SHARP:http://ng1.17img.cn/bbsfiles/images/2014/03/201403301059_494675_1733989_3.png为什么选择CD-BASEWAX气相毛细管柱做4-甲基咪唑就很好了呢?大家讨论一下!
共聚物氢谱为什么不符合n+1规律
求文献:乙二胺与乙酸反应制备2-甲基咪唑啉
请问食品中4-甲基咪唑用GCMS怎样做?
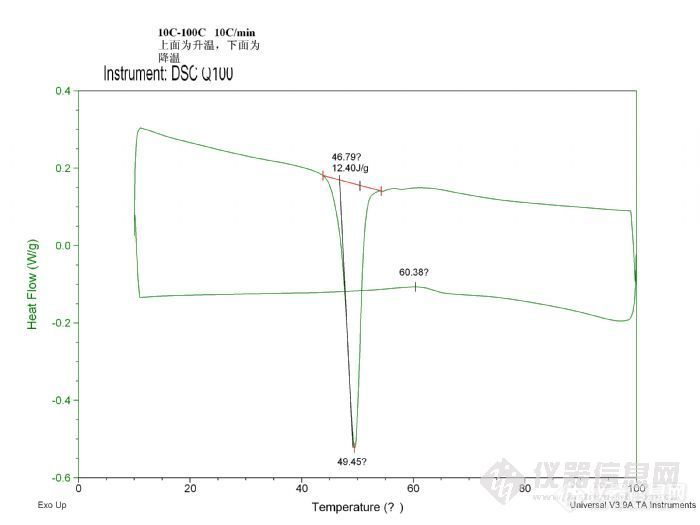
该样品为PCL与PB的嵌段共聚物,PCL的重量百分数为38%。下图给出的是一段升降温的DSC曲线图,奇怪的是降温过程没有出现明显的结晶特征峰,反倒是在60度左右出现了一个小的放热胞,该放热胞不可能是PCL结晶所致,是不是相转变过程导致的呢?还是其它原因所致。请各位同仁指点!http://ng1.17img.cn/bbsfiles/images/2010/09/201009011430_240356_2034213_3.jpg
默克密理博应用实验室 2013-07-15近日,百事可乐的产品在美国10个州中被爆出4-甲基咪唑(4-Methylimidazole)严重超标。4-甲基咪唑是一种有机中间体,主要用于合成大宗胃药西咪替丁,也可用作环氧树脂固化剂和金属表面防护剂等。可乐中的4-甲基咪唑是在以亚硫酸铵为原料生产焦糖色素时产生的。 4-甲基咪唑白色至类白色结晶粉末,易溶于水和乙醇,有腐蚀性,是一种能诱发肿瘤的化学物质。http://blog.merckmilliporechina.com/editor/upload/image/4C619C0F_7B615A74.PNG默克密理博致力于分析方法的开发,为客户提供简便、快速的解决方案。4-甲基咪唑及其异构体2-甲基咪唑均有较强极性,适合使用默克密理博的两性离子型亲水作用色谱柱(ZIC®-HILIC)分离。本实验采用默克密理博两性离子型(ZIC®-HILIC)色谱柱直接分析甲基咪唑的液相色谱方法。该方法前处理简单,不需要衍生化,也不需要添加离子对试剂。1 材料试剂1.1 对照品:4-甲基咪唑,2-甲基咪唑。1.2 色谱柱:ZIC®-HILIC 250-4.6mm 5um 200Å(默克密理博,货号:1.50458.0001)1.3 乙腈(默克密理博,货号:1.00030.4008)1.4 甲醇(默克密理博,货号:1.06007.4008)1.5 磷酸二氢钾(默克密理博,货号:1.04873.1000)1.6 可口可乐及百事可乐样品1.7 实验用为为超纯水(默克密理博Milli-Q Advantage)1.8 PVDF0.22um针头过滤器(默克密理博,货号:SLGV033NB)1.9 标准溶液配制:使用70%乙腈溶液,分别配制1mg/ml的4-甲基咪唑,2-甲基咪唑对照品原液。取两个对照品原液,1:1混合、稀释、定容,成,得100ug/ml的混合对照品母液。混合母液用70%乙腈溶液配制浓度为0.







 我要推广仪器
我要推广仪器
 下载APP
下载APP