食药总局:网络食安问题电商平台将承担连带责任
国家食品药品监督管理总局今日在北京召开新闻发布会,公布《网络食品安全违法行为查处办法》。据悉,该《办法》包括总则、网络食品安全义务、网络食品安全违法行为查处管理、法律责任、附则等,共五章48条,该办法将于2016年10月1日起实施。草酸二水合物 Oxalic acid dihydrate 6153-56-6双[3-(三乙氧基甲硅烷基)丙基]四硫化物 Bis[3-(triethoxysilyl)propyl] tetrasulfide 40372-72-3D-薄荷醇 D-Menthol 15356-60-2L-薄荷醇 L-Menthol 2216-51-51-十二烷醇 1-Dodecanol 112-53-81-十二烷醇 1-Dodecanol 112-53-81-十二烷醇 1-Dodecanol 112-53-81-辛醇 1-Octanol 111-87-55-甲基呋喃醛 5-Methylfurfural 620-02-0N-环己基甲酰胺 N-Cyclohexylformamide 766-93-84-甲基-2-戊醇 4-Methyl-2-pentanol 108-11-2N,N-二甲基-对苯二胺 N,N-Dimethyl-p-phenylenediamine 99-98-95,6,7,8-四氢-1-萘胺 5,6,7,8-Tetrahydro-1-naphthylamine 2217-41-6肼二盐酸盐 Hydrazine dihydrochloride 5341-61-7硫氰酸钾 Potassium thiocyanate 333-20-0二甲基硫醚 Dimethyl sulfide 75-18-3聚苯醚 Polyphenyl ether 31533-76-3叔丁基甲基醚 气相色谱级 Tert-Butyl methyl ether 1634-04-4七氟丁酸 Heptafluorobutyric acid 375-22-4甲苯二异氰酸酯 Tolylene Diisocyanate(TDI) 26471-62-53,4-二羟基苄胺氢溴酸盐 3,4-Dihydroxybenzylamine hydrobromide 16290-26-9N,N-二(羟基乙基)椰油酰胺 Coconut diethanolamide(CDEA) 68603-42-9/61791-31-9甲苯二异氰酸酯 Tolylene Diisocyanate(TDI) 26471-62-5异冰片基丙烯酸酯 Isobornyl acrylate 5888-33-5N,N' -二苯基硫脲 1,3-Diphenyl-2-thiourea 102-08-9聚合氯化铝 Aluminum chlorohydrate 1327-41-9四丁基氢氧化铵10%溶液 Tetrabutylammonium hydroxide solution 2052-49-5四丁基氢氧化铵25%溶液 Tetrabutylammonium hydroxide solution 2052-49-5L-苯基丙氨酸 L-Phenylalanine 63-91-2无水硫酸铈 Cerium(IV) sulfate 13590-82-4硫酸铈铵四水合物 Ammonium cerium(Ⅳ) sulfate tetrahydrate 18923-36-9脂蛋白脂肪酶 Lipoprotein Lipase 9004/2/8乙二胺≥99.5%标准品 Ethylenediamine 107-15-3壬二酸 Azelaic acid (Nonanedioic acid) 123-99-9N,N-二甲基-1-萘胺 N,N-Dimethyl-1-naphthylamine 86-56-6双(三氟甲烷)磺酰亚胺锂盐 Bis(trifluoromethane)sulfonimide lithium salt 90076-65-6




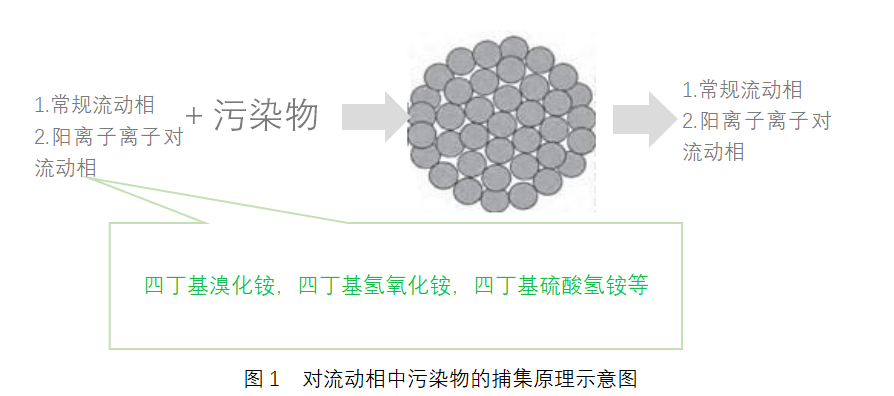























 我要推广仪器
我要推广仪器
 下载APP
下载APP