[color=#444444][color=#444444]求助,现在在做色谱检测这方面,要求分离苯甲酸,苯甲醛,苯甲醇,我用N-甲基吡咯烷酮作为内标物,溶剂为水。想问下N-甲基吡咯烷酮会与这三种物质发生反应吗?如果体系内有酸的情况下呢?[/color][/color]
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
[align=center][b]间三氟甲基苯丙醇和杂质I的分离[/b][/align]客户提供了间三氟甲基苯丙醇和相关杂质I,并反馈曾尝试使用反相C[sub]18[/sub]柱对两化合物进行分离,但未能得到基线分离结果。现客户希望本实验室选择合适色谱柱并对色谱条件进行优化,来实现间氟甲基苯丙醇和其相关杂质I的基线分离。首先,我们尝试使用中等极性的CAPCELLPAK C[sub]18[/sub] MGII色谱柱,在磷酸盐-乙腈体系中分析50 μg/mL的混标溶液及各单标溶液,通过调整流动相中水相和有机相比例为60:40时,50 μg/mL的混标溶液中,间三氟甲基苯丙醇和杂质I能实现基线分离,分离度为1.52(见图1)。同客户沟通,客户希望供试品溶液(当间三氟甲基苯丙醇浓度为1mg/mL,杂质I为1 μg/mL)中两化合物分离度大于1.50。[align=center][img=,422,132]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009027392_4941_2222981_3.png!w422x132.jpg[/img][/align][align=center][img=,656,427]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009243004_918_2222981_3.png!w656x427.jpg[/img][/align][align=center]图1 MGII分析混标及单标溶液结果[/align][align=left][img=,575,197]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031009245664_7431_2222981_3.png!w575x197.jpg[/img][/align][align=left]在此实验基础上,进一步分析供试品溶液,结果发现由于间三氟甲基苯丙醇浓度过高,致使色谱峰展宽,杂质I与间三氟甲基苯丙醇的分离度下降,未能达到1.50的基线分离要求;进一步尝试通过升高柱温来改善分离度,结果如图2,在50°C时能够得到良好分离结果,分离度为1.59。[/align][align=left][/align][align=center][img=,650,418]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031030364182_5088_2222981_3.png!w650x418.jpg[/img][/align][align=center]图2 MGII分析混标及单标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,575,195]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031031319132_5141_2222981_3.png!w575x195.jpg[/img][/align][align=left][/align][align=left]为有更多的选择,我们也尝试了两款非C[sub]18[/sub]色谱柱,包括键合特殊官能团——金刚烷基的高极性色谱柱ADME和键合五氟苯基的PFP色谱柱。在使用PFP色谱柱分析50 μg/mL混标溶液时,发现两化合物峰重合,未能实现分离。但使用ADME分析混标溶液时,能够得到1.36的分离度(见图3)。[/align][align=left][/align][align=center][img=,620,423]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034384978_3594_2222981_3.png!w620x423.jpg[/img][/align][align=center]图3 PFP、ADME分析50 μg/mL混标溶液结果[/align][align=left]注: 峰上标数字为分离度。[/align][align=left][img=,552,214]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031034366042_2199_2222981_3.png!w552x214.jpg[/img][/align][align=left][/align][align=left]尝试改善分离度,继续使用ADME色谱柱进行分析,通过降低有机相比例来延长保留,最终得到了1.50的分离度(见图4),与此同时对供试品溶液进行分析,发现由于主成分峰展宽未能得到基线分离结果(见图5)。[/align][align=left][/align][align=center][img=,658,430]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035399180_5905_2222981_3.png!w658x430.jpg[/img][/align][align=center]图4 ADME分析混标溶液结果[/align][align=center][/align][align=center][img=,657,435]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035148034_8911_2222981_3.png!w657x435.jpg[/img][/align][align=center]图5 ADME分析供试品溶液结果[/align]注: 峰上标数字为分离度。[align=left][img=,586,223]http://ng1.17img.cn/bbsfiles/images/2018/04/201804031035150115_8050_2222981_3.png!w586x223.jpg[/img][/align]
液相色谱峰刚开始有正峰和反峰连在一起,乙腈和水做流动相的,是正常的吗,分析的2.4.6-三甲基苯甲酰氯含量甲醇酯化做的
我想问下大家,我用三甲基硅烷基重氮甲烷衍生青霉素,然后加入甲醇做催化剂,空白里没有青霉素,可是从峰上看,出现苯环结构,想问问有没有大神知道原因呢,求告知
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
最近要做50325三苯的标曲,采购没搞清楚买了一套甲醇溶剂的标液,抱着侥幸心理实验了两天,还是拖尾严重,真是愁死了。仪器某国产二次热解析,岛津2014c,成本原因只能用活性炭管,各位大佬有没有解决方法啊?
[size=18px]目前在用AB的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测三苯基氯甲烷,Q1 MI模式扫243.1的离子[font=-apple-system, BlinkMacSystemFont, &](应该是三苯甲基碳正离子)[/font],发现基线非常高(30万-50万之间),且不稳定,时高时低,导致峰面积也 不稳定,打电话问客服,几个人几种说法,“液相部分污染了”“这个是正常现象,多走走就稳定了”,尝试用MRM模式去做,打出一个165.2的碎片,基线不到1000,做了线性和回收也都挺好,但是,这个碎片离子是怎么打出来的比较困惑,就怕以后再做的时候重现不出来……[/size][size=18px]流动相是90%甲醇,溶剂是正丁醇:乙腈(80:20)[/size][size=18px]请教一下各位大神,AB的仪器用SIM模式选择Q1 MI还是Q3 MI好呢?基线高且时高时低,除了污染还有什么原因呢?[font=-apple-system, BlinkMacSystemFont, &]三苯甲基碳正离子在质谱里能被打碎吗?会裂解成什么碎片离子?[/font][/size][size=18px][font=-apple-system, BlinkMacSystemFont, &][/font][/size]
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
我用的是FID检测器,在一台岛津GC-2014C上同时有甲醇(溶剂是水)和三苯(溶剂二硫化碳),我们的A瓶是二硫化碳清洗剂,B瓶是水清洗剂,C瓶是丙酮清洗剂。最开始没放丙酮,先走甲醇后又走三苯,空白都走的两遍,但是一直堵针,于是就在甲醇和三苯之间走了丙酮洗针,结果导致二硫化碳空白一直有干扰峰,反过来,就是水空白有很多杂峰,(有丙酮峰也有其他峰),而且空白走三次都没用。我是新手,想问问大家怎么处理,还是水的和二硫化碳的不能在一台仪器上走哪?
求救:请问那位大师有[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定三苯、山梨酸、苯甲酸、甲醇的不确定度分析啊??我们下周就要实验室认可了,急需!谢谢!
买来的甲醇中甲基汞是69.5ug/g,若要配成中间储备液如何配?个人觉得是69.5*0.7918(甲醇密度)=55.03ppm 再进行稀释 但是身边有同事提出需要换算到汞的含量再稀释即 69.5/215.63(甲基汞摩尔质量)*201(汞摩尔质量)*0.7918(甲醇密度)=51.30ppm 现在思路比较乱,还请论坛上大神帮帮忙!万分感谢~
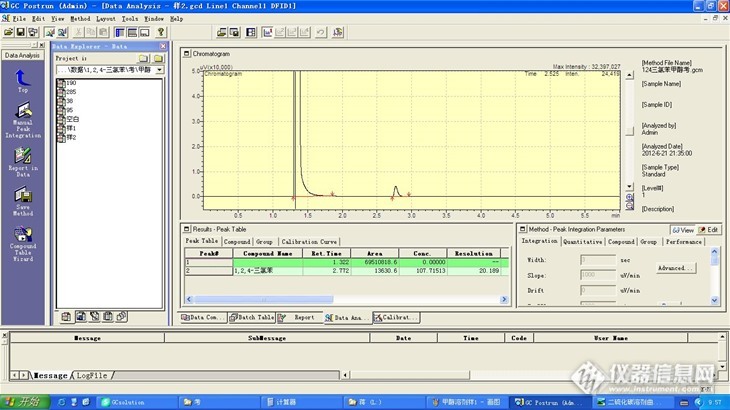
经常在论坛看见有人发帖问同一种物质在不同溶剂(讨论最多的是二硫化碳和甲醇)做标准曲线对分析结果的影响。我就以上次专家来考核甲醇中1,2,4-三氯苯盲样为例讨论下分流进样时,分别用二硫化碳和甲醇做溶剂对峰面积、保留时间的影响。 公司扩项中有环境空气和废气中1,2,4-三氯苯。方法是大气固定污染源 氯苯类化合物的测定 气相色谱法 HJ/T66-2001,专家来考核的时候带来了甲醇中1,2,4-三氯苯盲样。于是问题来了:标准里是用二硫化碳做溶剂的而盲样是以甲醇做溶剂,能用二硫化碳溶剂做的曲线来标定盲样吗?我先后用二硫化碳和甲醇做溶剂来试验。 实验部分仪器和试剂岛津气相2014C色谱仪带AOC20i自动进样器,FID检测器。1,2,4-三氯苯(色谱纯)二硫化碳(色谱纯)甲醇(色谱纯) 色谱条件毛细柱OV-101 25m*0.20mm*0.25um柱箱温度140℃,进样口温度250℃,FID检测器温度250℃,氢气40ml/min,空气400ml/min,氮气30cm/s;进样量1ul,分流比50。盲样注明考核浓度范围为1.0-150.0mg/L所以配制二硫化碳中1,2,4-三氯苯浓度分别为(ug/ml) 0,20,40,80,100,200 甲醇中1,2,4-三氯苯浓度分别为(ug/ml) 0,38,95,190,285http://ng1.17img.cn/bbsfiles/images/2013/10/201310121116_470549_2103464_3.jpg这是二硫化碳溶剂空白http://ng1.17img.cn/bbsfiles/images/2013/10/201310121116_470550_2103464_3.jpg这是甲醇溶剂空白,有拖尾。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121118_470551_2103464_3.jpg这是二硫化碳做溶剂标液100ug/mlhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121121_470553_2103464_3.jpg这是甲醇做溶剂标液95ug/mlhttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121122_470554_2103464_3.jpg这是二硫化碳做溶剂的曲线http://ng1.17img.cn/bbsfiles/images/2013/10/201310121123_470555_2103464_3.jpg这是甲醇做溶剂的曲线,第一个点空白为零但是空白的数据方框不能选中(谁能解释下?这可能是不强制过零点所致),这样其实和没带入空白的曲线是一样的。有没代入空白的零点计算对截距有点影响。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121132_470559_2103464_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121133_470560_2103464_3.jpg这是盲样做的两针平行样加载二硫化碳溶剂曲线结果分别为 94.0ug/ml ,92.6ug/ml 平均值93.3ug/ml。这个结果偏小不合格。http://ng1.17img.cn/bbsfiles/images/2013/10/201310121138_470561_2103464_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310121138_470562_2103464_3.jpg这是盲样做的两针平行样加载甲醇溶剂曲线结果分别为 109.4ug/ml ,107.7ug/ml 平均值108.6ug/ml。这个结果是合格的。再看下保留时间:二硫化碳做溶剂时1,2,4-三氯苯保留时间为2.783 ,甲醇做溶剂时1,2,4-三氯苯保留时间为2.772,几乎一样。结论:用二硫化碳做溶剂1,2,4-三氯苯的峰面积明显大于以甲醇做溶剂时的峰面积,所以以二硫化碳做溶剂的曲线来测以甲醇做溶剂的样品结果会偏小。这可能和不同溶剂在气化室状态不同有关,而保留时间几乎不变。
我想了解4-氯-2-三氟甲基苯腈(4-Chloro-2-(trifluoromethyl)benzonitrile,CAS#320-41-2)的理化性质,但在网上只找到沸点109 º C (10 mmHg),是液体还是固体看不出来。因为这个沸点是真空条件下的。那位老师有相关的信息,请告诉我,谢谢!
各位高手上午好,最近我们要做18581-2009中三苯及甲醇的测试,在看完标准后我遇到以下问题,希望有高手指点下。1、该标准是用内标法定量,说要称取与待测物式样中同一数量级的内标物和样品,可是限量里面它们的数量级都不一样,这要怎么办呢?2、标准里规定的已苯和二甲苯的限量是求和,那我在做校正因子的时候是不是也可以将它们的面积加到一起算呢?3、我用的气象色谱型号为GC1690,灵敏度很低,而我们用的试剂又是分析纯的,经常会在目标物附近出小峰(极性柱PEG20M和非极性柱SE-54都会有),我要怎么判断这些小峰是不是目标物呢?
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。
什么样的填充柱对甲醇吸附较大,苯分离效果显著 我想分析甲醇中的苯系物,甲醇拖尾太大,无法精确定量.请教高人指导.
高手们帮帮我吧。。苦恼了一个多月了。我用的安捷伦7820,做苯系物。只要测苯,甲苯,乙苯,邻间对二甲苯。。。用的甲醇做溶剂。热解析进样。用的强极性柱子DB-WAXETR 30米*0.32毫米*1.0微米条件是 进样口:150度,分流10:1(20:1和40:1都试过) 恒流模式:3.0mL/min 柱温:程序升温50度保持7分钟,10度每分钟升到110,保持5分钟 检测器:250度 尾吹30mL/min买的甲醇中苯系物的标准样品1mg/mL,配成的100微克/毫升,50,20,10,5,0……的浓度,进1微升结果除了甲醇和苯出峰超差,别的都分开,三个在一起的峰勉强分的还行吧。关键溶剂峰甲醇峰拖尾很厉害,在拖尾上就出苯峰了,而且苯峰特别小,50浓度的时候就已经很小了,再低浓度已经没法做了。。上个关于甲醇和苯的出峰图,高手们帮我看看吧。
请问各位大虾:结晶紫指示液、萘酚苯甲醇指示液、甲基橙的饱和丙酮溶液、高氯酸滴定液的保存期限各是多少?谢谢了
热解析法测甲醇中苯系物,直接进样的方式进标样出峰正常,通过吸附管进样,三连峰分不开,是什么原因,该怎么解决呢?
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
大家好,是否有一种好的采样技术,可以同时富集废气中的苯胺、硝基苯、苯、氯苯、甲醇等组分,然后方便直接用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析定量?不知道用固相微萃取技术能不能实现呀?
用的FFAP毛细管柱,甲醇为空白溶剂,苯峰在甲醇峰的拖尾处,乙醇与苯峰又很近。当乙醇含量高时又会积分成苯峰。用什么极性的柱子可以分离这三个峰呀。
苯环上3位和5位上各带一个三氟甲基,扫碳谱后非常的杂,如何才能比较准确地找出相对应的峰并计算耦合常数呢?三氟甲基上的碳是否裂分位一个4重峰了,临位的碳也被裂分为双峰了呢?
各位高手,小弟我最近要接手师姐的课题开始做实验了,我们要测鱼油里面的脂肪酸。师姐给我的方法里面有一个试剂是14%三氟化硼甲醇溶液,我知道这个东西要用三氟化硼和甲醇配,我们实验室有一瓶很旧的三氟化硼,打开瓶子的时候吓我一跳:冒了好多烟,闻起来胸闷,我怕是这个东西有毒就不敢用了。师姐说可以去买别人配好的三氟化硼甲醇,比较安全,但是我找不到哪里有卖的,西格玛也说没有这个东西,有没有哪位之前买过这个东西的,告诉我哪里可以买到啊,进口国产都无所谓,只要质量好就行了!PS:还有个问题,脂肪酸甲酯化用甲醇就好了,为神马还要三氟化硼啊?三氟化硼分子上没有甲基啊?
今天甲醇中苯的标准曲线,标准溶液是1ug/ul的,进样体积是0.1,0.2,0.5,1,2,做完前三个点还是准的,第四个点开始峰面积偏小,做了很多次都是,小弟是新手,求高手支招。另外进样的浓度需要按照标准上来吗?稀释的话该怎么稀释,因为这次专家来看,求大神指点迷津。本人用的岛津GC-2014C
[size=4][font=宋体]求香菇上氟乐灵,甲基毒死蜱,毒死蜱,氯杀螨,乙硫磷,氯苯嘧啶醇[/font][/size][size=4][font=Times New Roman] [/font][/size][size=4][font=宋体]氟虫腈的前处理方法,能够有效去除杂质的,谢谢大家,这个香菇上的杂质太多了,用弗洛里硅土不管用,杂质很多。[/font][/size]
有的书上说,三氟甲基在碳谱中可产生1:3:3:1的四重峰,距离比较远。本人做过一次,基本上只得到两个较高峰,两边的小峰有扫不出来的可能吗?而且,三氟甲基对alfa碳有裂分吗?
用7820A来做苯系物的标线,用的是甲醇中的9种苯系物标液,柱子是DB-WAX,60米,0.25mm的柱子,要分离甲醇跟苯的条件是什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP