最多用30ML乙酸丁酯萃取过2.5g镓,但不知道最大量是多少?有人知道乙酸丁酯能萃取镓的最大量吗?谢谢
[color=#444444]有没有人做食品中兽残丙酸睾酮的测定?[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法测定时,色谱柱中一直有残留,洗不下来,有没有好的方法?我参考的是SN/T3235-2012,流动相用的0.1%甲酸水溶液和0.1%甲酸甲醇溶液[/color]
乙酸乙脂萃取金时,如果金量太多,乙酸乙脂会从水相上坠到水相下.这样不便于分液,所以大家一定要注意金的称样量.[em0814]
求教一下做过乙酸乙酯萃取纯金然后测杂质含量的朋友,含有1mol/L盐酸的乙酸乙酯怎么配制?
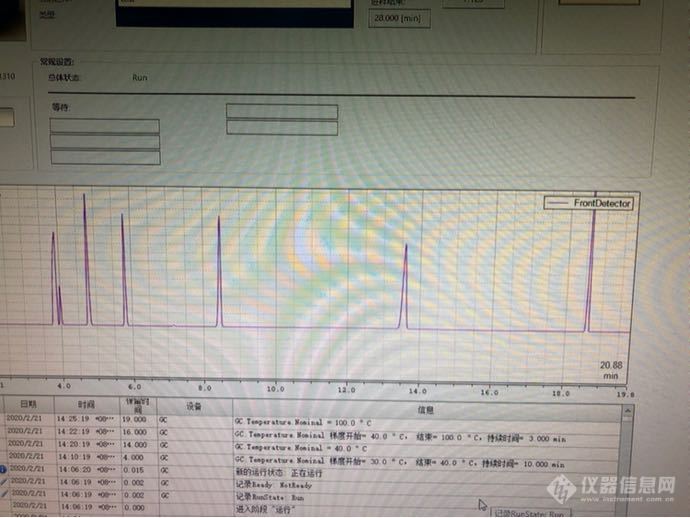
我买的坛墨的7种饱和脂肪酸酯类化合物混标!甲酸甲酯 乙酯 乙酸甲乙丙丁戊酯。现在问题来了。我甲酸乙酯和乙酸甲酯一直分不开 。下面是我条件:30 度保持4min 以1 度/min 升到40度 30度/min到100。后面都没问题 甲酸甲酯我温度低一点也能和cs2分开。可是甲酸乙酯和乙酸甲酯...分不开。那我怎么做呢?是不是要单独做一个乙酸甲酯曲线方法验证。柱子是 ffap 30 0.32 0.25[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/02/202002211425479329_4319_2990176_3.png[/img]
我今天按照国标GB/T5750.10-2006中的9测定二氯乙酸,进行的是加标回收。因为是用甲基叔丁基醚做的介质,质谱分析时有很多峰,我无法确定哪一个是二氯乙酸甲酯,自带的质谱库内也没有。请问哪位能帮我找一下二氯乙酸甲酯的质谱分析参数,谢谢。
乙酸甲酯里面有微量醋酸,有什么好的检测方法吗
上层为乙酸乙酯,下层为2mol/L的盐酸溶液,震摇萃取后,乙酸乙酯层乳化,有什么好的办法,不影响结果的情况下,快速破乳嘛?[img]https://ng1.17img.cn/bbsfiles/images/2023/03/202303141332180134_7311_3304234_3.png[/img]
目前我有一个体系中含有甲醇,乙酸,乙酸甲酯,以及少量的硫酸,我想问怎么才能够准确的知道甲醇,乙酸,乙酸甲酯的量,另外气相在进样过程中如何避免甲醇和乙酸之间的反应
大家好,我现在在做乙酰乙酸甲酯气相色谱检测,用同一根色谱柱进样,有时候时两个峰,有时候是一个峰到底是什么原因?乙酰乙酸甲酯有互变异构体,在运行过程中有出现两个峰的可能。 现在能找到乙酰乙酸甲酯和乙酰乙酸乙酯的国标。乙酰乙酸甲酯的国标中是一各峰,而乙酰乙酸乙酯是两个峰,这两个东西的性质是相似的,为什么会出现不同的峰形呢?
请教一下做过用乙酸乙酯萃取法,测高纯金杂质含量的朋友,我做回收的时候Sn的回收率总是很低,然后我对萃取后的乙酸乙酯蒸发后测杂质,没有发现缺少的Sn?这是什么情况
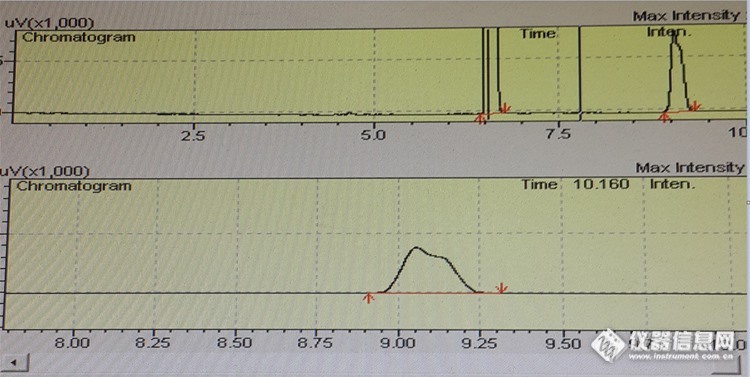
职业卫生 乙酸甲酯与甲酸乙酯同事用HP-INNOWAX 试过了说分不开,这次我用DB-FFAP来试试也分不开。因为聚乙二醇类柱子在职业卫生比较好用,哪种聚乙二醇柱子可以分开乙酸甲酯与甲酸乙酯?我的色谱条件 DB-FFAP(30m*0.53mm*1.0um) 柱温40℃ 线速度12cm/s 分流比50 检测器 进样器均为200℃http://ng1.17img.cn/bbsfiles/images/2016/05/201605051534_592369_2103464_3.png
用乙酸丁酯萃取矿石中的金,上原子吸收火焰法测定,没有吸光度啊。为什么呢?原吸的参数都调好了呀。
用乙酸乙酯做有机氯标曲,低浓度的从10、20、40、50、100、200,中间浓度的是20、40、80、100、200、400,高浓度的是 100、200、400、500、1000、2000ppb,标曲线性不好,原因何在?ECD检测器,tr-1色谱柱。
做偶氮二异丁腈,发现有资料用乙酸甲酯:水=1:1做流动相,反相色谱,所以想问问乙酸甲酯是否可以用于反相色谱?乙酸乙酯是用于正相色谱,所以求助
[color=#444444]本人最近按2015版药典做了一个药用辅料-醋酸羟丙甲纤维素琥珀酸酯的的游离乙酸的含量测定实验。实验过程如下:[/color][color=#444444] 游离乙酸、琥珀酸 取本品0.102g,精密称定,置锥形瓶中,精密加入磷酸盐溶液(取0.02mol/L磷酸二氢钾溶液,用1mol/L氢氧化钠溶液调pH值至7.5)4.0ml,搅拌2小时,加磷酸溶液(取1.25mol/L磷酸1ml,置50ml量瓶中,加水稀释至刻度,摇匀)4.0ml,强力振摇,离心,上清液作为供试品溶液;精密称取琥珀酸0.13g,置100ml量瓶中,加水适量,振摇使完全溶解,加水至刻度,摇匀,作为琥珀酸贮备液;取加有水20ml的100ml量瓶,称重,精密加入冰乙酸2ml,再称重,用水稀释至刻度,摇匀,精密量取6ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为乙酸贮备溶液;精密量取乙酸贮备液和琥珀酸贮备液各4.0ml,置同一25ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。照高效液相色谱法(中国药典2015年版四部通则0512)试验。以十八烷基硅烷键合硅胶为填充剂,以0.02moI/L磷酸二氢钾溶液(用6mol/L磷酸溶液调pH值至2.8)为流动相,流速每分钟1ml,检测波长为215nm。取对照溶液10μl, 注入液相色谱仪,按琥珀酸峰计算,理论板数不得少于8000。取供试品溶液与对照溶液各10μl,注入液相色谱仪,按干燥品计算,游离乙酸和琥珀酸总量不得过1.0%。[/color][color=#444444]计算公式: 游离乙酸含量=0.0768(WA/(W(1-干燥失重)))(γUA/γSA)[/color][color=#444444] 式中 WA为乙酸贮备溶液中冰乙酸量,mg;[/color][color=#444444] W为供试品的取样量,mg;[/color][color=#444444] γUA、γSA为供试品溶液、对照溶液中乙酸的峰面积。[/color][color=#444444] 游离琥珀酸含量=1.28(WS/(WUS(1-干燥失重)))(γUS/γSS)[/color][color=#444444] 式中 WS为琥珀酸贮备液中琥珀酸量,mg;[/color][color=#444444] WUS为供试品取样量,mg;[/color][color=#444444] γUS、γSS为供试品溶液、对照溶液琥珀酸的峰面积。[/color][color=#444444]我的问题是,根据“干燥品计算,游离乙酸和琥珀酸总量不得过1.0%”这句话,游离乙酸含量的最后计算的结果要不要乘以100%,比如我最后计算结果是0.0139,如果这个结果再乘以100%,就变为1.39%,从而超过限度,那么就需要重新做实验复核一遍。[/color]
刚收到的乙酸甲酯样品:什么信息都没有,只是用GC做了纯度一项,其它的还要分析哪些项目。哪位有乙酸(醋酸)甲酯的检测方法或标准或者有相关分析经验的,可否拿出分享,不胜感谢!

前几天同事扩项工作场所空气中饱和脂肪酸酯类物质包括乙酸甲酯,乙酸乙酯,乙酸丙酯,乙酸丁酯,标准是GBZ/T160.63-2007。柱子是SH-Rtx5(30m*0.32mm*0.25um),同事欲恒温同时分离这四种酯类,我提示乙酸乙酯,乙酸丙酯,乙酸丁酯混一起恒温做没事,如果乙酸甲酯也混一起做那么会与溶剂二硫化碳峰难分离,于是他计划先做乙酸乙酯,乙酸丙酯,乙酸丁酯再另外单独做乙酸甲酯。 乙酸乙酯,乙酸丙酯,乙酸丁酯色谱条件:岛津气相色谱GC2010PLUS 柱温60℃ 检测器进样器均为200℃ 分流比50 恒线速度22cm/shttp://ng1.17img.cn/bbsfiles/images/2017/01/201701191702_673938_2103464_3.jpg三种乙酸酯峰型还不错。接下来他说想试试乙酸甲酯在这个条件下出峰会怎么样?因为我之前用OV101做过二硫化碳中乙酸甲酯,它是紧挨着在二硫化碳前出峰,同时也用DB-FFAP做过它是在二硫化碳之后。SH-Rtx5极性比OV101强些 比DB-FFAP弱很多,那么在二硫化碳之前还是二硫化碳之后出峰呢? 看到甲乙丙丁突然有了一想法:不是有碳数规律吗?利用碳数规律推测乙酸甲酯的保留时间:保留时间:乙酸乙酯 2.854min 乙酸丙酯 3.562min 乙酸丁酯5.162min 二硫化碳2.612min碳数规律:lnt‘=An+C t’为调整保留时间 n为同系物中碳个数 A, C均为常数首先精确计算死时间:间隔均匀同系物 精确死时间计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616020890_01_2103464_3.pngtm=(2.854*5.162-3.562*3.562)/2.854+5.162-3.562-3.562=2.292min调整保留时间代入碳数规律公式:乙酸乙酯ln0.562=4A+C 乙酸丙酯 ln1.27=5A+C 求得乙酸甲酯调整保留时间lnt’=3A+C t’=0.249min乙酸甲酯的预测保留时间0.249+2.292=2.541min.这个保留时间在二硫化碳(2.612min)之前,两者仅仅相差0.07min。于是预测同条件下乙酸甲酯在二硫化碳之前出峰并且分离度不好!同条件实验做二硫化碳中乙酸甲酯3000ug/ml来验证:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616132954_01_2103464_3.jpg实验结果乙酸甲酯保留时间是2.571min与预测的2.541min比较符合,外推是有误差的,而且本例碳数不多,碳数多些会更准确。降低柱温至40℃,线速度15cm/s 乙酸甲酯与二硫化碳分离达到定量要求!http://ng1.17img.cn/bbsfiles/images/2016/09/201609061616_608604_2103464_3.jpg 结论:碳数规律还是比较准的,可以预测出峰情况。
我使用固相萃取仪(SPE4790)萃取完后,萃取液如何去除水和酸,达到什么要求才能进样,参考方法是552.1。萃取出来的物质时卤乙酸酯吗?
摘 要 本文以去离子水为萃取剂,通过加电膜萃取装置萃取了乙酸丁酯中的无机阴离子。在600V直流电压的作用下,8 ml乙酸丁酯中的四种无机阴离子通过聚丙烯中空纤维膜孔内的有机液膜进入膜内的100 μl 去离子水中。萃取完成后,使用离子色谱对萃取液进行分析。本文优化了影响萃取过程的因素,如施加电压、搅拌速度以及萃取时间。最佳萃取条件为:施加电压为600V,搅拌速度为600rpm,萃取时间为5分钟。该方法成功应用于乙酸丁酯实际样品的测定,无机阴离子的线性范围为0.01 mg/L-1mg/L,加标回收率为97-102%。实验结果表明,该方法快速有效,并可应用于其他微溶性有机物中无机阴离子的测定。关键词 无机阴离子;乙酸丁酯;加电中空纤维膜萃取;离子色谱 最近15年来,中空纤维膜萃取技术作为一种样品前处理方法,被广泛应用于环境分析、药物分析以及食品和饮料检测等重要领域。中空纤维膜萃取技术具有样品无需处理即可直接进样,对分析物具有预富集作用,基体消除,成本低廉,易与气相色谱、高效液相色谱、毛细管电流和离子色谱联用等优点。但分析物穿透中空纤维膜壁上的支撑液膜的运输机理是基于被动扩散,因此,常常需要花费超过30分钟甚至长达一小时的时间才能达到萃取平衡。为了克服传统的中空纤维膜萃取技术耗时的缺点,2005年挪威的Pedersen-Bjergaard和Rasmussen首次提出了一种称为加电中空纤维膜萃取的新型样品前处理方法。短短5分钟内,碱性药物在300V直流电压的作用下能够成功地穿过一层薄薄的有机液膜进入到300 μl 接收相中。与传统基于被动扩散的膜萃取技术相比,电动力迁移在加电中空纤维膜萃取中起到了主导作用,因此在很短的时间内就能取得良好的萃取效果。随后,Pedersen-Bjergaar小组又将这种新型的加电中空纤维膜萃取方法应用于更多种类的碱性药物、酸性药物、肽类和氯酚类物质的萃取 。他们建立了加电中空纤维膜萃取过程的理论模型并考察了影响加电中空纤维膜萃取效率的参数 。此外,新加坡的Lee小组使用加电中空纤维膜萃取技术从羊水、血清、口红和尿样中萃取了铅离子。目前为止,加电中空纤维膜萃取技术一直应用于液-液-液三相萃取体系,而本实验首次将加电中空纤维膜萃取应用于液-液两相萃取体系。我们曾经使用在线中空纤维膜萃取-离子色谱法测定了乙酯乙酯的无机阴离子,但该方法的萃取时间长达半小时,并且使用了柱切换技术,装置复杂,操作繁琐,仪器成本高。而本实验使用加电中空纤维膜萃取技术仅需要短短5分钟就能完成萃取,并且装置简单,操作简便,成功应用于乙酸丁酯中无机阴离子的测定。
活性炭管同时采了甲苯与乙酸甲酯,二硫化碳解吸。甲苯我一直用非极性柱做,乙酸甲酯极性柱做。这要分两次进样麻烦。岛津2014c自动进样那台装的是OV101非极性柱。我想用OV101柱(25m*0.20mm*0.25um)同时做甲苯和乙酸甲酯。最大的麻烦是二硫化碳与乙酸甲酯分离的问题,有谁成功的用非极性柱分离过二硫化碳溶剂峰和乙酸甲酯吗?
[color=#444444]甲酰基苯乙酸甲酯的色谱含量测试中。由于甲酰基苯乙酸甲酯会有醇酮互变性质,在液相色谱(乙腈:水=50:50)下出现三个峰。但是在LC——MS下从第一个峰开始到第三个峰这之间所有的时间都出现了M+1峰(包括峰之间的)。谁能告诉我这是怎么回事。[/color][color=#444444]有哪个大侠做过甲酰基苯乙酸甲酯的含量测试的,求指导。[/color]
大家好,我急求用岛津气相色谱分离,乙酸和乙酸乙酯的分离条件,柱子,进样口,检测器温度,以及乙醇和乙酸乙酯的分离条件,柱子采用RTX-17ms,急求,谢谢大家!!!
大家好,有人做过土壤对乙酸乙酯的吸附实验吗?吸附等温线是怎么做的,不同浓度的乙酸乙酯怎么配制的,是配制在甲醇还是什么溶剂里,土壤应加多少量?另外,平衡溶液中水中的乙酸乙酯怎么萃取,主要是用什么溶剂来萃取,因为要用GC-FID来测定其含量,所以乙酸乙酯里面不能有水,谢谢!
一般的扩项需要跑标曲但是GB/T 10345-2007白酒分析方法中对乙酸乙酯,乳酸乙酯这些没有明确标曲标液点取样量,我想问一下大家都是怎么扩白酒这些项目的,是保持内标物添加量不变,直接增大目标物标液量吗?[img]https://ng1.17img.cn/bbsfiles/images/2018/10/201810311129322310_5286_3302856_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/10/201810311129321629_7570_3302856_3.png[/img]
最近没有事,常想一个问题,我们做金中杂质分析时用乙酸乙酯,而很多单位却用乙醚,大家能告诉我吗?我试着比较两种物质.对比如下1\安全性不用说乙醚危险,极易燃烧,属于管制药品.而乙酸乙酯安全的多,而且满大街都有卖的.2\操作乙醚在萃取过程 ,分层很慢,有的单位做试样时,需要静止5分钟以上,而乙酸乙酯只需要静止半分钟就完全能分离.3\效果乙醚萃取时,如果时间不够长,通常水相颜色很深(萃取不完全),而乙酸乙酯不出现这个问题.4\纯化 乙醚的沸点低,在34度就能进行蒸馏,而乙酸乙酯纯化稍微麻烦,但是在78度蒸馏产物完全能达到萃取效果,而且其中杂质含量和乙醚蒸馏后大致相当.5\金回收由于乙醚沸点较低,按理将其蒸发很容易,然而温度太高,乙醚易着火燃烧,而使金损失严重,另外乙醚直接排出也不利于环保,所以只能通过蒸馏来回收乙醚并分析金.但是在该过程中,蒸馏反应剧烈,可能造成喷溅.相比而言,乙酸乙酯由于沸点高,所以直接在电热板上加热除去乙酸乙酯即可.从上面我觉得应该选择乙酸乙酯,而大多数单位却选择了乙醚,这是为什么呢?
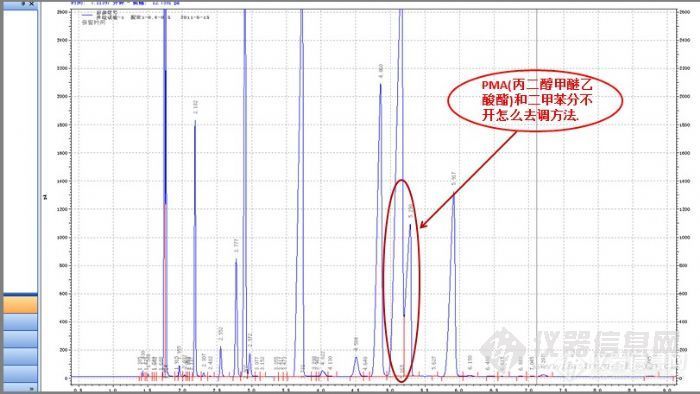
测涂料里的苯系物 PMA+二甲苯分不开各位: 1、我今天做了一下PU白面漆的苯系物含量,但做出来的谱图里面的 PMA(丙二醇甲醚乙酸酯)和那个二甲苯分不开,要怎么去调方法呢? 2、做涂料的苯系物含量是不是要先按要求配比好(漆:固化剂:稀释剂),再加一点乙酸乙脂稀释,再离心一下,取上部清液分析呢。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191653_630872_2263490_3.jpg
我们生产工业乙酸乙酯,乙醇和乙酸酯化得到的。中控分析用的是TCD(因为要分析出水分含量)。做校正因子的时候遇到以下情况第一种方法:买的试剂(无水乙醇,乙酸乙酯分析纯)因为含有水分所以我用分子筛吸附了水分,吸附好以后在库仑水分仪上测得水分0.001%。然后在天平上配置标液。做校正因子,用校正面积归一法定量第二种方法:买的试剂不经分子筛去水直接配置标液做校正因子,用校正面积归一法定量上述两种定量方法测得的乙醇相对乙酸乙酯的校正因子差了很多。第一种测得是f=1.6,第二种0.7,查文献是0.8..想不通什么原因,难道是分子筛处理的原因。请专家指教。谢谢
如何将丙酮、乙酸甲酯、石油醚分开???
用WXF-310型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计测金 ;做不出线性,50%王水容样在水浴上煮。放置澄清,吸取上成清夜。加水乙酸丁脂有机相萃取测定金。求助相同的方法做的好的。谢谢、、、、急急急







 我要推广仪器
我要推广仪器
 下载APP
下载APP