糖苷酶抑制剂标准品哪里找?上海甄准生物
糖苷酶抑制剂标准品哪里找?------上海甄准生物 糖苷酶抑制剂是一类含氮的拟糖类结构能抑制糖苷键形成的化合物。从结构上可分为两组:第一组氮原子在环上有野尻霉素(nojirimycin)、半乳糖苷酶抑素(galactostatin)、寡糖酶抑素(oligostatin)等。第二组氮原子在环外,如阿卡糖(acarbose),validoxylamine A、B,有效霉素A、B(海藻糖苷酶抑制剂)等,从抑制酶范围上看,它包括了部分&alpha -葡萄糖苷酶抑制剂、半乳糖酶抑制剂、唾液酸抑制剂、淀粉酶抑制剂。 上海甄准生物提供糖苷酶抑制剂标准品,为您检测分析提供强有力支持! 产品信息: 货号 品名 CAS No. B691000 N-Butyldeoxynojirimycin Hydrochloride 210110-90-0 C10H22ClNO4 10/100mg a-葡糖苷酶1和 HIV cytopathicity抑制剂 E915000 N-Ethyldeoxynojirimycin Hydrochloride 210241-65-9 C8H18ClNO4 10/100mg HIV cytopathicity抑制剂 C181150 N-5-Carboxypentyl-deoxymannojirimycin 104154-10-1 C12H23NO6 5/50mg 制备亲和树脂的配体,用于纯化Man9 甘露糖苷酶 A187545 2,3-O-Acetyloxy-2&rsquo ,3&rsquo ,4&rsquo ,6,6&rsquo -penta-O-benzyl-4-O-D-glucopyranosyl N-Benzyloxycarbonylmoranoline (&alpha /&beta mixture) C56H63NO13 10/100mg 4-O-&alpha -D-Glucopyranosylmoranoline 制备中间体 B690500 N-(n-Butyl)deoxygalactonojirimycin 141206-42-0 C10H21NO45/50mg a-D-半乳糖苷酶抑制剂 B690750 N-Butyldeoxymannojirimycin, Hydrochloride 355012-88-3 C10H22ClNO4 5/50mg a-D-甘露糖苷酶抑制剂 D236000 Deoxyfuconojirimycin, Hydrochloride 210174-73-5 C6H14ClNO3 10/100mg alpha-L-岩藻糖苷酶抑制剂 M166000 D-Manno-&gamma -lactam 62362-63-4 C6H11NO5 5/50mgalpha-甘露糖苷酶 ß - 葡糖苷酶抑制剂和 M165150 D-Mannojirimycin Bisulfite C6H13NO7S 1/10mg alpha-甘露糖苷酶抑制剂 D455000 6,7-Dihydroxyswainsonine 144367-16-8 C8H15NO5 1/10mg a-甘露糖苷酶抑制剂 C665000 Conduritol B 25348-64-5 C6H10O4 25/250mg b-葡糖苷酶抑制剂 C666000 Conduritol B Epoxide 6090-95-5 C6H10O5 25/250mg b-葡糖苷酶抑制剂 A155250 2-Acetamido-2-deoxy-D-gluconhydroximo-1,5-lactone 1,3,4,6-tetraacetate 132152-77-3 C16H22N2O10 25/250mg glucosamidase抑制剂 D240000 Deoxymannojirimycin Hydrochloride 73465-43-7 C6H14ClNO4 10/100mg mammalian Golgi alpha- mannosidase 1 抑制剂 M297000 N-Methyldeoxynojirimycin69567-10-8 C7H15NO4 10/100mg N-连接糖蛋白高斯过程干扰剂 A158400 2-Acetamido-1,2-dideoxynojirimycin 105265-96-1 C8H16N2O4 1/10mg N-乙酰葡糖胺糖苷酶抑制剂 A157250 O-(2-Acetamido-2-deoxy-D-glucopyranosylidene)amino N-Phenylcarbamate 132489-69-1 C15H19N3O7 5/10/100mg O-糖苷酶,己糖胺酶A和己糖胺酶B抑制剂 A157252 (Z)-O-(2-Acetamido-2-deoxy-D-glucopyranosylidene)amino N-Phenyl-d5-carbamate 1331383-16-4 C15H14D5N3O7 1/10mg O-糖苷酶,己糖胺酶A和己糖胺酶B抑制剂 M334515 4-Methylumbelliferyl &alpha -D-Glucopyranoside 4&rsquo -O-C6-N-Hydroxysuccinimide Ester C26H31NO12 25mg T2DM糖苷酶抑制剂 G450000 4-O-&alpha -D-Glucopyranosylmoranoline 80312-32-9 C12H23NO9 1/10mg &alpha -葡萄糖苷酶抑制剂 D231750 1-Deoxy-L-altronojirimycin Hydrochloride 355138-93-1 C6H14ClNO4 5/50mg &alpha -糖苷酶抑制剂 H942000 N-(2-Hydroxyethyl)-1-deoxy-L-altronojirimycin Hydrochloride Salt C8H18ClNO5 0.5/5mg &alpha -糖苷酶抑制剂 H942015 N-(2-Hydroxyethyl)-1-deoxygalactonojirimycin Hydrochloride C8H18ClNO5 1/10mg &alpha -糖苷酶抑制剂 H942030 N-(2-Hydroxyethyl)-1-deoxy-L-idonojirimycin Hydrochloride C8H18ClNO55/50mg &alpha -糖苷酶抑制剂 T795200 3&rsquo ,4&rsquo ,7-Trihydroxyisoflavone 485-63-2 C15H10O5 200mg/2g &beta -半乳糖苷酶抑制剂 A158380 O-(2-Acetamido-2-deoxy-3,4,6-tri-o-acetyl-D-glucopyranosylidene)amino N-(4-nitrophenyl)carbamate 351421-19-7 C21H24N4O12 10/100mg 氨基葡萄糖苷酶抑制剂 M166505 Mannostatin A, 3,4-Carbamate 1,2-Cyclohexyl Ketal C13H19NO4S 2.5/25mg 保护的Mannostatin A B682500 Bromoconduritol (Mixture of Isomers) 42014-74-4 C6H9O3Br 200mg 哺乳类 alpha-葡萄糖苷酶 2 抑制剂 K450000 Kifunensine 109944-15-2 C8H12N2O6 1/10mg 芳基甘露糖苷酶抑制剂 D239750 1-Deoxy-L-idonojirimycin Hydrochloride 210223-32-8 C6H14ClNO4 10/100mg 酵母葡糖a-苷酶类抑制剂S885000 Swainsonine 72741-87-8 C8H15NO3 1/10mg 可逆,活性部位直接抑制甘露糖苷酶抑制剂;Golgi a-甘露糖苷酶 II抑制剂 T295810 [1S-(1&alpha ,2&alpha ,8&beta ,8a&beta )]-2,3,8,8a-Tetrahydro-1,2,8-trihydroxy-5(1H)-indolizinone 149952-74-9 C8H11NO4 10/100mg 苦马豆素和衍生物合成中间体 N635000 Nojirimycin-1-Sulfonic Acid 114417-84-4 C6H13NO7S 10/100mg 葡糖苷酶类抑制剂 V094000(+)-Valienamine Hydrochloride 38231-86-6 C7H14ClNO4 1/10mg 葡糖苷酶抑制剂 D440000 2,5-Dideoxy-2,5-imino-D-mannitol 59920-31-9 C6H13NO4 1/10mg 葡糖苷酶抑制剂 D494550 N-Dodecyldeoxynojirimycin 79206-22-7 C18H37NO4 10/100mg 葡糖苷酶整理剂 D479955 2,4-Dinitrophenyl 2-Deoxy-2-fluoro-&beta -D-glucopyranoside 111495-86-4 C12H13FN2O9 5/50mg 葡糖基氟化物,可以作为特定的机制为基础的糖苷酶抑制剂,未来可应用于合成和降解的低聚糖和多糖 A653270 2,5-Anhydro D-Mannose Oxime, Technical grade 127676-61-3 C6H11NO5 10/100mg 潜在的葡苷糖酶抑制剂C-(D-吡葡亚硝脲)乙胺和C-(D-glycofuranosyl)甲胺 D236500 1-Deoxygalactonojirimycin Hydrochloride 75172-81-5 C6H14ClNO4 10/100mg 强效的和有选择性的d半乳糖苷酶抑制剂 D236502 Deoxygalactonojirimycin-15N Hydrochloride C6H14Cl15NO4 5/25mg 强效的和有选择性的d半乳糖苷酶抑制剂 B445000 (2S,5S)-Bishydroxymethyl-(3R,4R)-bishydroxypyrrolidine 105015-44-9 C6H13NO4 10/100mg 强有力的和特定的糖苷酶抑制剂 M166500 Mannostatin A, Hydrochloride 134235-13-5 C6H14ClNO3S 1/10mg 强有力的糖苷酶抑制剂,甘露糖苷酶抑制剂 A858000 N-(4-Azidosalicyl)-6-amido-6-deoxy-glucopyranose 86979-66-0 C13H16N4O7 1/10mg 人类红细胞单糖运输标签抑制剂 C185000 Castanospermine 79831-76-8 C8H15NO4 10/100mg 溶酶体 a-或者beta-葡糖苷酶. 葡糖苷酶1抑制剂和 beta-甘露糖苷酶抑制剂 D439980 1,4-Dideoxy-1,4-imino-D-mannitol, Hydrochloride 114976-76-0 C6H14ClNO4 5/50mg 糖蛋白甘露糖苷酶抑制剂 A608080 N-(12-Aminododecyl)deoxynojirimycin 885484-41-3 C12H26N2O4 5/50mg 糖苷酶亚氨基糖醇制备用试剂 I866350 1,2-O-Isopropylidene-alpha-D-xylo-pentodialdo-1,4-furanose 53167-11-6 C8H12O5 100mg/1g 糖苷酶抑制剂制备试剂 A648300 2,5-Anhydro-2,5-imino-D-glucitol 132295-44-4 C6H13NO4 10/100mg 糖水解酶类抑制剂 A648350 2,5-Anhydro-2,5-imino-D-mannitol 59920-31-9 C6H13NO4 1/10mg 糖水解酶类抑制剂 M257000 3-Mercaptopicolinic Acid Hydrochloride 320386-54-7 C6H6ClNO2S 500mg/5g 糖质新生抑制剂 B286255 N-Benzyloxycarbonyl-4,6-O-phenylmethylene Deoxynojirimycin 138381-83-6 C21H23NO6 5/50mg 脱氧野尻霉素衍生物 B286260 N-Benzyloxycarbonyl-4,6-O-phenylmethylene Deoxynojirimycin Diacetate 153373-52-5 C25H27NO8 2.5/25mg 脱氧野尻霉素衍生物 D245000 Deoxynojirimycin 19130-96-2 C6H13NO4 10/100mg 脱氧野尻霉素抑制哺乳类葡糖苷酶1 A172200 N-Acetyl-2,3-dehydro-2-deoxyneuraminic Acid Sodium Salt 209977-53-7 C11H16NNaO8 10/100mg 细菌、动物和病毒抑制剂 C181200 N-5-Carboxypentyl-1-deoxynojirimycin 79206-51-2 C12H23NO6 5/50mg 制备亲和树脂的配体,用于纯化葡糖苷酶I C181205 N-5-Carboxypentyl-1-deoxygalactonojirimycin 1240479-07-5 C12H23NO6 5/50mg 制备亲和树脂的配体,用于纯化葡糖苷酶I C645000 Conduritol A 牛奶菜醇A 526-87-4 C6H10O4 1/10mg C667000 Conduritol D牛奶菜醇D 4782-75-6 C6H10O4 10mg I868875 1,2-Isopropylidene Swainsonine 85624-09-5 C11H19NO31/10mg 更多产品,更多优惠!请联系我们! 上海甄准生物科技有限公司 免费热线:400-002-3832










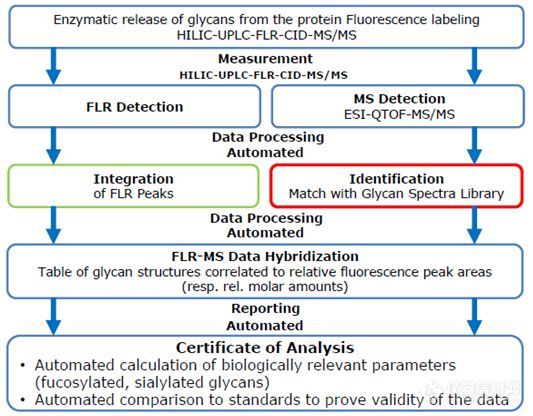







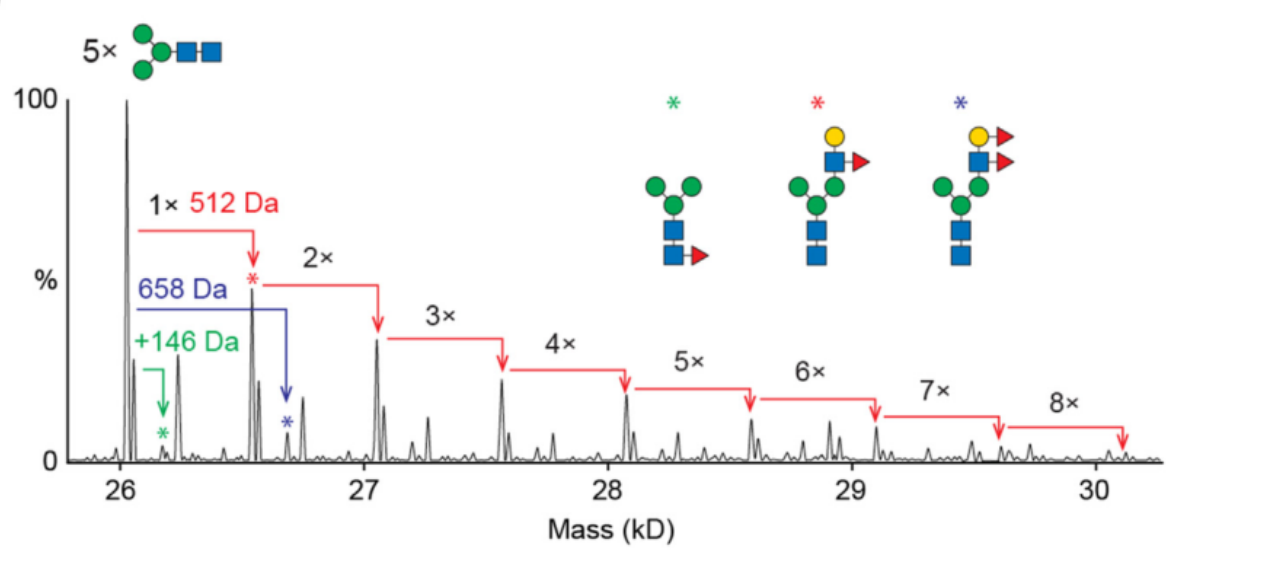








 我要推广仪器
我要推广仪器
 下载APP
下载APP