各位大神,哪有有二正辛基-双(巯乙酸2-乙基己酯)(DOTE)和三(2-乙基己基巯基乙酸)辛锡(MOTE)反应物料,或者有卖三(2-乙基己基巯基乙酸)辛锡(MOTE)的供应商啊。网上百度,相关信息为零。这是SVHC中的一项物质。http://simg.instrument.com.cn/bbs/images/default/em09512.gif
大家好,请问氯乙酸、巯基乙酸、巯基丙酸(含量都比较高80%以上)能用液相色谱来检测吗?如果能的话用什么样的色谱柱、什么色谱条件,谢谢了
谁告诉我下 巯基乙酸异辛酯 二甲基二氯锡 用GC什么检测器?什么柱子分析啊?
GB/T5750-2006中,用铬天青S法测水中铝时,掩蔽铁锰的掩蔽剂是巯基乙醇酸。可试剂供应商说没有巯基乙醇酸,只有巯基乙酸。现求助各位专家,巯基乙醇酸溶液怎么配制?
目前在检测巯基乙酸和巯基丙酸,想将两者分开,目前找不到合适的毛细柱,大家有什么好办法吗?
大家有做过这个巯基乙酸的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]验证吗,溶剂是乙腈:水=1:9。然后就发现空白也有值,就考虑试剂干扰,分析后发现乙腈没有影响,而水居然有干扰,想知道大家做的时候出现过这种情况吗,流动相为0.1%甲酸水和乙腈流动相。如果我把溶剂换成纯乙腈,巯基乙酸能溶吗,这会影响到样品的提取吗
各位老师们,有人用离子色谱测试过化妆品中的巯基乙酸吗?如果有经验不妨拿出来分享一下啊!附件里面的文献重复不出来呢。
化妆品中巯基乙酸的检测方法(征求意见稿)1 范围 本方法规定了离子色谱法测定化妆品中巯基乙酸的含量。 本方法适用于化妆品中巯基乙酸及其盐类和酯类含量的测定。2 方法提要样品中的巯基乙酸经水溶解提取后,用离子色谱仪分离巯基乙酸根与无机离子,电导检测器检测,以保留时间定性,峰面积定量。本方法巯基乙酸的检出限5.8ng,定量下限20ng。取样量为0.5g时,检出浓度为46μg/g,最低定量浓度0.15mg/g。3 试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T6682规定的一级水。3.1 巯基乙酸,优级纯。3.2 甲醇,优级纯。3.3 三氯甲烷,分析纯。3.4 氢氧化钠,分析纯。3.5 硫酸溶液:取硫酸(ρ20=1.84g/ml)10mL,缓慢加入到90mL水中,混匀。3.6 盐酸溶液:取盐酸(ρ20=1.19g/ml)10mL,加入90mL水中,混匀。3.7 淀粉溶液(10g/L):称可溶性淀粉1g,加水5mL调成溶液,再加入沸水95mL,煮沸,并加水杨酸0.1g或氯化锌0.4g防腐。3.8 氢氧化钠溶液(500g/L):称取氢氧化钠50g,加水适量使溶解并至100mL。再量取一定量用经超声脱气的水稀释到淋洗液浓度。3.9 重铬酸钾标准溶液:准确称取已于120℃±2℃电烘箱中干燥至恒重的重铬酸钾基准物质4.9031g,溶于水并转移至1000mL量瓶中,定容至刻度,摇匀。3.10 硫代硫酸钠标准溶液(0.1mol/L):称取硫代硫酸钠(Na2S2O3▪5H2O)26g(或无水硫代硫酸钠16g)溶于1000mL新煮沸放冷的水中,加入氢氧化钠0.4g或无水碳酸钠0.2g,摇匀,贮存于棕色瓶内,放置两周后过滤,用重铬酸钾标准溶液标定其浓度,标定方法如下:准确吸取重铬酸钾标准溶液(3.9)25.00mL于500mL碘量瓶中,加碘化钾2.0g和硫酸溶液(10%)20mL,立即密塞,摇匀,于暗处放置10min。加入水150mL,用硫代硫酸钠标准溶液滴定至溶液呈淡黄色时,加入淀粉溶液(3.7)2mL,继续滴定至蓝色变为亮绿色。同时做空白试验。按下式计算硫代硫酸钠标准溶液的浓度。c(Na2S2O3)=Cx25.0/(v1-v0) 式中:c(Na2S2O3)——硫代硫酸钠标准溶液的浓度,mol/L; C——重铬酸钾标准溶液的浓度,mol/L; V1——硫代硫酸钠标准溶液的体积,mL; V0——空白试验硫代硫酸钠溶液的体积,mL。3.11 碘标准溶液(0.05mol/L):称取碘13.0g和碘化钾35g,加水100mL使溶解,加入盐酸3滴,用水稀释至1000mL,过滤后转入棕色瓶中。3.12 巯基乙酸标准储备溶液(10g/L):称取巯基乙酸标准品(3.1)1.0g,用水稀释并转移至100mL容量瓶中,加入甲醛1mL,加水定容得标准储备溶液,临用时标定。标定方法如下:准确吸取巯基乙酸标准储备溶液10.00mL,置于250ml碘量瓶中,加入水10mL,盐酸10mL,精确加入碘标准溶液(3.11)20.00mL,混匀,于暗处放置3分钟,用硫代硫酸钠标准溶液滴定,至溶液颜色变为浅黄色时,加入淀粉溶液(3.7)2mL,(溶液变为蓝色,)继续滴定至蓝色消失即为终点。同时做空白试验,按下式计算巯基乙酸标准溶液的浓度。c(HSCH2COOH)=92.1xcx(v0-v1)X1000X1000/(VX1000 )式中:c(HSCH2COOH)——巯基乙酸标准储备溶液的浓度,μg/mL; C——硫代硫酸钠标准溶液的浓度,mol/L; V0——空白试验消耗硫代硫酸钠标准溶液的体积,mL; V1——巯基乙酸标准储备溶液消耗硫代硫酸钠溶标准液的体积,mL; V——巯基乙酸标准储备溶液的体积,mL; 92.1——巯基乙酸的摩尔质量,g/mol。4 仪器和设备4.1离子色谱仪,抑制型电导检测器。4.2 电子天平。4.3 涡旋振荡器。4.4 超声波清洗器。4.5 高速离心机。5 分析步骤5.1 巯基乙酸标准系列溶液制备临用新配。取经碘量法标定的巯基乙酸标准储备溶液适量,分别配制成0.50 mg/L、1.00 mg/L、2.00 mg/L、5.00 mg/L、10.0 mg/L、20.0 mg/L、50.0 mg/L、80.0mg/L的巯基乙酸标准系列溶液。5.2样品处理称取样品0.5g(精确到0.001g)于100mL具塞比色管中,加水至刻度,膏状样品用涡旋振荡器振摇均匀,超声波清洗器提取20min,加入三氯甲烷(3.3)2mL,轻轻振摇,静置。浑浊样品,在14000rpm转速下高速离心15min,取上清液经0.25μm滤膜过滤,滤液作为待测溶液。5.3 参考色谱条件 色谱柱:AS11-HC(250×4mm I.D.),AG11-HC(50×4mm I.D.),柱填料为强碱性离子交换树脂,烷醇季铵作功能基;抑制器:ASRS ULTRA;抑制模式:外接水1.0mL/min,自动抑制电流50mA;淋洗液: 25mmol/LNaOH+1%甲醇混合液;淋洗液流速:0.85mL/min;柱温:室温;进样量:25μl;5.4 测定在“5.3”色谱条件下,取巯基乙酸标准系列溶液(5.1)分别进样,记录色谱图,以标准系列溶液为横坐标,峰面积为纵坐标,绘制标准曲线。取“5.2”项下的样品待测溶液进样,记录色谱图,量取峰面积,根据标准曲线得到样品待测溶液中巯基乙酸的浓度。按“6”计算样品中巯基乙酸的含量。6 计算 按下式计算巯基乙酸的浓度(以巯基乙酸计): ω(巯基乙酸)=BXV/m 式中:ω(巯基乙酸)– 样品中巯基乙酸的质量分数,μg/g; B– 测试溶液中巯基乙酸的质量浓度,mg/L; V– 样品定容体积,mL; m– 样品取样量,g。7 色谱图 http://ng1.17img.cn/bbsfiles/images/2014/12/201412050826_525963_1680138_3.gif图1 巯基乙酸标准溶液离子色谱图1:巯基乙酸(TR=16.587)
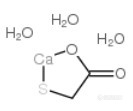
巯基乙酸钙盐三水合物 CAS号:5793-98-6 分子式:C2H8CaO5S 分子量 184 结构式http://ng1.17img.cn/bbsfiles/images/2017/10/2016042817011772_01_1490617_3.png 《化妆品安全技术规范》(2015年版)当中,3.9巯基乙酸第三法——化学滴定法的反应方程如下:https://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_670059_1490617_3.png 原理是https://ng1.17img.cn/bbsfiles/images/2016/04/201604281715_591808_1490617_3.png 该方法的适用范围中这样描述:本方法适用于脱毛类、烫发类和其他发用类化妆品中巯基乙酸及其盐类和酯类含量的测定。客户委托了一款产品,要求按照巯基乙酸钙含量出报告,含量计算公式中有一个系数0.184,描述是1mmol碘溶液相当于巯基乙酸钙的克数,这样显然其指的巯基乙酸钙不是CAS:814-71-1 分子式C4H6CaO4S2(分子量222.3),不知道巯基乙酸钙盐三水合物是否依然按照上述原理与碘反应。 求高手指教,前辈指点!谢谢
不知道大家有没有做过染发剂中巯基乙酸的测定呢?是化妆品卫生规范2007中的第二法化学滴定法,感觉终点很难判断,因为染发剂本来有颜色,怎么样更好的判断是否达到终点?
化妆品中巯基乙酸的检测方法
请高手指教!!!巯基乙醇酸和巯基乙酸是同一种试剂吗?如果不是,它还有别的名称吗?哪里能购到?
给位大侠,我在根据国标法测奶粉中的牛磺酸时,配置乙酸锌溶液用来去蛋白,可是乙酸锌不溶于水。通过加热溶解时发现40℃时几乎没溶,后来水浴温度调到80℃时不停搅拌后还有一些未溶,但是温度再高的话里面的水又会蒸发了,于是又加了点凉水进去,随着温度的降低乙酸锌又被析出来了,放置一夜后烧杯上全是乙酸锌,而且烧杯里面乙酸锌又与原先加水时的量一样多,这是怎么回事?
做油过氧化值时,配置的乙酸异辛烷发生了分层现象。乙酸异辛烷的比例是按照国标GB/T 5538 中规定的3:2配置的。不知道大家有遇到过这种情况吗?是什么原因导致分层呢?
我是做蛋白质SDS-PAGE电泳的。蛋白质样品用碘乙酸溶液处理后如何除去过量的碘乙酸?1、取约1 mg的总蛋白样品放入离心管中,加入1ml 6M 的尿素溶液,得到1 mg/mL的蛋白溶液。2、加入20微升1M的巯基乙醇到上述离心管中,震荡混合5秒钟。3、将上述离心管恒温在37oC条件下反应1小时 (以还原蛋白中的双硫键)。4、加入25µ L 1M新鲜配制在1M NaOH中的碘乙酸,在避光、室温条件下放置30 分钟。(烷基化将巯基保护起来) 接下来我准备做SDS-PAGE还原电泳,我不清楚样品中的过量碘乙酸对电泳有没有影响?应该怎么除掉?求高人指点![em0808][em0808][em0808][em0808][em0808]
配置流动相,没有氯化锌,可以用乙酸锌代替吗

http://ng1.17img.cn/bbsfiles/images/2015/06/201506170935_550426_2770921_3.jpg乙酸锌-乙酸钠配置出来是浑浊的,想问问大家配出来是怎么样的,求图
大家好,我急求用岛津气相色谱分离,乙酸和乙酸乙酯的分离条件,柱子,进样口,检测器温度,以及乙醇和乙酸乙酯的分离条件,柱子采用RTX-17ms,急求,谢谢大家!!!
5750.10-2023新国标二氯乙酸和三氯乙酸的结果计算公式,不打算用方法给的公式,还有其他推出来的公式吗,求指教[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211154108_8493_6020871_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211155038_7049_6020871_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307042211155782_3893_6020871_3.png[/img]
如何定性及定量过氧乙酸和过氧辛酸,有过氧乙酸对照品,无过氧辛酸对照品.两种物质都比较容易分解.有经验或有方法的发出来共享一下.谢谢大侠!
提前感谢各位赐教。看氰化物的标准时发现,5750中氰化物(异烟酸-吡唑酮法)蒸馏时使用的是乙酸锌溶液+固体酒石酸,而484氰化物(异烟酸-吡唑啉酮)中用到的则是硝酸锌溶液和酒石酸溶液。想问一下这两者的的区别在哪里呢?日常操作中是否可以用乙酸锌溶液代替硝酸锌溶液呢?
想请教一下各位老师,为什么我用C18柱检测一针乙酸乙酯的时候,为什么乙酸乙酯在2min就出峰了,相似相容的话,乙酸乙酯应该最迟出峰啊?
在用铬天青S法做水中铝中,用到的 巯基乙醇酸溶液(10g/L)是如何配制的,请知道的同志给予帮助解答,非常感谢!
要分析乙酸中的甲酸和乙酸乙酯,TCD检测器,填充柱。 用GDX加酸不太好分离,请教诸位同仁有无经验?
各位老师,乙酰乙酸乙酯说有烯醇类和酮类平衡的混合物。那是不是就是说,我进一针乙酰乙酸乙酯的分析纯,应该出两个峰,一个乙酰乙酸乙酯(烯醇类),一个是乙酯乙酯乙酯(酮类)。
[color=#444444]GC-2014C[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]做标准曲线,TCD(热导)检测器。标准溶液是水、乙醇、乙酸和乙酸乙酯的混合物,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]出的图中,水合乙醇的峰很好,但乙酸和乙酸乙酯的峰有问题,乙酸的峰拖尾严重,乙酸乙酯的峰很小(质量分数和水差不多,但峰面积比水差很多,有事几乎没有)。进样器140℃,柱温120℃,检测器130℃(以前三个温度分别是175,140,175但是这种条件下乙酸乙酯量很少时15%以下,不出峰)。请问有没有办法是避免乙酸峰拖尾太严重,另外增加乙酸乙酯的峰面积?[/color]
铬天青光度法测饮用水中铝GB/T5750,对高铁样品标准上上说用2ml巯基乙醇酸10g/L的可消除,可是买不到这个巯基乙醇酸试剂,是不是标准错了,还是有别名?
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
新国标5750.10中的二氯乙酸回收率为什么很高,超出回收率范围,求大佬指教
各位老师:HPLC色谱条件下进样乙酸乙酯,分别在磷酸二氢钾(ph3.0)和甲酸流动相体系下乙酸乙酯的响应值相差较大,是否有同样的问题出现?







 我要推广仪器
我要推广仪器
 下载APP
下载APP