请教大家我用国标方法做4-甲基咪唑,回收率只有20%是什么原因?我的过程是:取15mL5mg/L的4-甲基咪唑标准品,加1mol/L的碳酸钠5mL, 混匀,加入20mL80:20的三氯甲烷和乙醇混合物,摇荡提取,弃去水层,再用20mL50mM的甲烷磺酸液提取,过滤上机,可是为什么回收率这么差呢?
大家好: 我现在有个离子液体样品,里面含有大概1%的N-甲基咪唑量,因为用气相色谱做这个样品对柱子有损坏作用,现在想通过液相色谱法对N甲基咪唑做定量分析。我公司有安捷伦的液相色谱,柱子是C18的,检测器有紫外和视差检测器。我们自己通过液相实验后分析不出样品中的N甲基咪唑,但进纯样N甲基咪唑后有信号峰出现。想求教下大家有什么好方法能分析出离子液体样品中的N甲基咪唑。
有网友问:做4-甲基咪唑不出峰。如何解决。
急需以下化学试剂标准四氢呋喃、吗啉、DMF-DMA、N-甲基哌嗪、甲磺酸、对甲苯磺酸有知道的同志请将结果发送至我的邮箱weiweidou2008@live.cn谢谢大家啦!

2-甲基咪唑: 分子式 CH3C3H3N2 , 分子量 82.1048, CAS号 693-98-1 性质 固体。熔点142~143℃。沸点267~268℃。溶于水、乙醇、微溶于冷苯。分子式如下:http://ng1.17img.cn/bbsfiles/images/2012/01/201201091849_344855_2357013_3.jpg问题:大家有没有用气相色谱仪测试2-甲基咪唑的方法,或者给出一些相关建议也行,集思广益,希望大家多多发表间接。我自己的做法如下,样品:分析纯2-甲基咪唑0.07g溶于20ml纯水,用FID测试,进样口温度270、柱箱200、检测器270,测试的结果不理想,图谱如下:第一个峰单独进空气验证排除空气峰的可能性,可能是2-甲基咪唑中的杂质峰;第二个峰应该是进样针中残留的2-甲基咪唑,按照此图谱是不是说明可以确认2-甲基咪唑的含量?有版友说此结果只能说明是GC测试的含量,因为样品可能含有一些胺盐FID上是无法响应的,另外2-甲基咪唑含有叔胺和仲胺,这个在FID上的响应是不是也会低一些?http://ng1.17img.cn/bbsfiles/images/2012/01/201201091902_344856_2357013_3.jpg
今年3月,CSPI发布报告称,**可乐等4种产品中,均发现了一种名为4-甲基咪唑的化学物质,应当用何种检测方法?
焦糖色中4-甲基咪唑的测定是采用薄层色谱法,可是最近我在测定样品时,根据标准方法进行操作,却没有标准样品点却没有显色。不知道哪位同仁做过这个检测,烦请帮忙!

前不久安谱技术接到一个售前,客户想做4-甲基咪唑,用了HP-5 的柱子 分离情况谱图如下:http://ng1.17img.cn/bbsfiles/images/2014/03/201403301052_494672_1733989_3.jpg用HP-5分离情况不好,安谱的工程师给客户选择了CD-BASEWAX 谱图情况如下,出峰很SHARP:http://ng1.17img.cn/bbsfiles/images/2014/03/201403301059_494675_1733989_3.png为什么选择CD-BASEWAX气相毛细管柱做4-甲基咪唑就很好了呢?大家讨论一下!
求文献:乙二胺与乙酸反应制备2-甲基咪唑啉
请问食品中4-甲基咪唑用GCMS怎样做?
一下子买不到色谱纯或分析纯的甲磺酸,于是就用了化学纯的甲磺酸,只是背景稍高于以前,分离效果目前也没有什么问题,不知道以后会不会有影响。
默克密理博应用实验室 2013-07-15近日,百事可乐的产品在美国10个州中被爆出4-甲基咪唑(4-Methylimidazole)严重超标。4-甲基咪唑是一种有机中间体,主要用于合成大宗胃药西咪替丁,也可用作环氧树脂固化剂和金属表面防护剂等。可乐中的4-甲基咪唑是在以亚硫酸铵为原料生产焦糖色素时产生的。 4-甲基咪唑白色至类白色结晶粉末,易溶于水和乙醇,有腐蚀性,是一种能诱发肿瘤的化学物质。http://blog.merckmilliporechina.com/editor/upload/image/4C619C0F_7B615A74.PNG默克密理博致力于分析方法的开发,为客户提供简便、快速的解决方案。4-甲基咪唑及其异构体2-甲基咪唑均有较强极性,适合使用默克密理博的两性离子型亲水作用色谱柱(ZIC®-HILIC)分离。本实验采用默克密理博两性离子型(ZIC®-HILIC)色谱柱直接分析甲基咪唑的液相色谱方法。该方法前处理简单,不需要衍生化,也不需要添加离子对试剂。1 材料试剂1.1 对照品:4-甲基咪唑,2-甲基咪唑。1.2 色谱柱:ZIC®-HILIC 250-4.6mm 5um 200Å(默克密理博,货号:1.50458.0001)1.3 乙腈(默克密理博,货号:1.00030.4008)1.4 甲醇(默克密理博,货号:1.06007.4008)1.5 磷酸二氢钾(默克密理博,货号:1.04873.1000)1.6 可口可乐及百事可乐样品1.7 实验用为为超纯水(默克密理博Milli-Q Advantage)1.8 PVDF0.22um针头过滤器(默克密理博,货号:SLGV033NB)1.9 标准溶液配制:使用70%乙腈溶液,分别配制1mg/ml的4-甲基咪唑,2-甲基咪唑对照品原液。取两个对照品原液,1:1混合、稀释、定容,成,得100ug/ml的混合对照品母液。混合母液用70%乙腈溶液配制浓度为0.
现在我们要用气相检测甲磺酸中的甲磺酸甲酯和甲磺酸乙酯,不知道甲磺酸能不能直接进样? 对柱子和仪器有什么要求不?感谢
有报道称可口可乐有极微量的致癌物质4-甲基咪唑,(美国355毫升可口可乐中每kg含量为4微克,中国为56微克,英国为135微克),不会对人体有什么影响。请问可乐样品如何来用GCMS测定4-甲基咪唑?用什么样品处理方法?
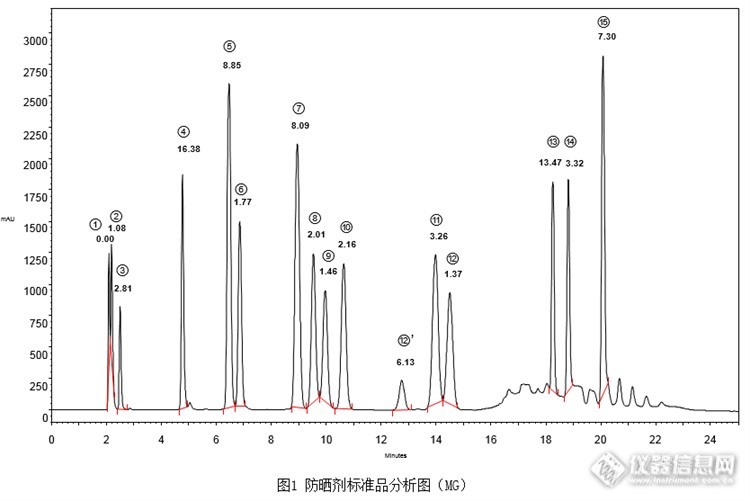
[align=center][b]2015版《化妆品安全技术规范》防晒剂检验方法-苯基苯并咪唑磺酸等15种组分[/b][/align][align=center][b]第一法(高效液相色谱-二极管阵列检测器法)[/b][/align]本次实验按照2015版《化妆品安全技术规范》中防晒剂检验方法的第一法(高效液相色谱-二极管阵列检测器法),对苯基苯并咪唑磺酸等15种防晒剂进行同时分析。15种防晒剂标准品按照《化妆品安全技术规范》配制成混合标准溶液,分别使用CAPCELL PAK C18 MG S5 4.6 mm i.d. × 250 mm,CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,CAPCELL PAK ADME S5 4.6 mm i.d. ×250 mm,CAPCELL PAK C18 AQ S5 4.6 mm i.d. × 250 mm以及SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm五款色谱柱对混合标准溶液进行分析。其中,MG和MGII色谱柱得到相对较好结果,但两款色谱柱原流动相条件下,个别峰未实现基线分离。结果如图1、图2。[img=,690,460]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_01_2222981_3.png[/img][img=,690,432]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_02_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,304]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_03_2222981_3.png[/img]为得到更好的分离效果,使用1支更新的MGII色谱柱,在原流动相条件基础上,对梯度进行调整,结果如图3所示。各峰分离度得到明显改善,但峰11和峰12分离度为1.43,仍未达到基线分离。[img=,690,425]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,292]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_02_2222981_3.png[/img]继续调整梯度条件,分析结果如4所示。在此条件下,各峰实现基线分离,得到良好分析结果。[img=,690,421]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170935_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,307]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170937_01_2222981_3.png[/img]接下来将色谱柱更换为MG色谱柱,在调整后的梯度条件下进行分析,结果如图5所示,同样可得到良好的分析结果。[img=,690,419]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170938_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,291]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170940_01_2222981_3.png[/img]
求助三氟甲磺酸酐气相测试方法我们用HP-5测试 不知道测试出来的是不是主峰,并且杂质较多,求助测试方法?是否可以做硅化测试?
2-甲基苯并咪唑和邻硝基苯胺的的紫外吸光度事多少?
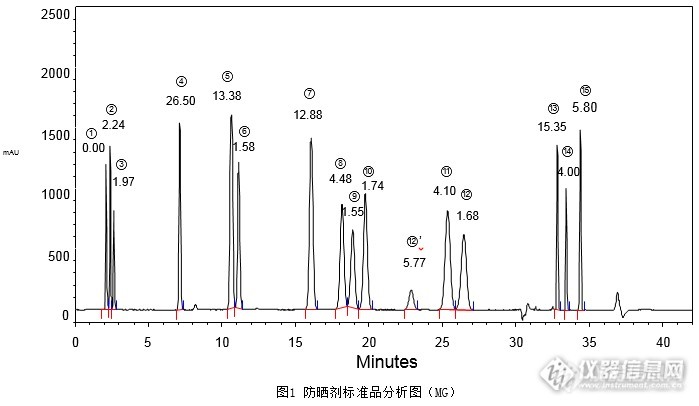
[align=center][b]2015版《化妆品安全技术规范》防晒剂检验方法[/b][/align][align=center][b]苯基苯并咪唑磺酸等15种组分-二元梯度法[/b][/align][align=center][b] [/b][/align]在2015版《化妆品安全技术规范》防晒剂检验方法中,第一法对15种防晒剂的分析为三元梯度方法,此法要求仪器配备三元泵,且四氢呋喃会对PEEK基材的管路和仪器有溶胀作用,所以该方法在实际操作上会受到一定的限制;而第二法将15种防晒剂分为两组,在不同流动相条件下分别检测,较为费时费力,且前12种防晒剂峰未能达到基线分离。基于以上情况,本次实验采用二元梯度方法,对15种防晒剂标准品进行同时分析,既可在常规二元泵系统进行实验,也可免去分组分析的繁琐。本实验混合标准溶液按照《化妆品安全技术规范》配制,分别使用资生堂CAPCELLPAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 250 mm和CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d.× 250 mm色谱柱进行分析,结果如图1和图2所示,两款色谱柱在二元梯度条件下均可使15种防晒剂峰实现基线分离。[img=,690,400]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_01_2222981_3.png[/img][img=,690,366]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_02_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,629,207]http://ng1.17img.cn/bbsfiles/images/2017/08/201708230913_03_2222981_3.png[/img]
急需甲磺酸丁酯,哪位大虾可以指点从哪里可以购买到,急急!
GB/T29668-2013化妆品用防腐剂 双(羟甲基)咪唑烷基脲
美国一家机构检测可口可乐,发现含有致癌物质4-甲基咪唑(简称:4-MEI),这种与啮齿动物患肺癌有关的化学物质被曝出现在可乐配方焦糖色素中。 因为涉及到饮料业巨头,此事一经曝光,引起广泛关注。 我国的“中国饮料工业协会”也发表声明,表明立场,全文如下: 为提高饮料产品的食品安全性、及时与企业、媒体及消费者保持沟通,我协会一直高度关注“焦糖色中4甲基咪唑”的有关动态,就目前我协会掌握和了解的情况,我国饮料中所含的焦糖色不会致癌,可安全饮用。具体说明如下: 1、无论国外还是国内,焦糖色被允许用于饮料等食品。在我国焦糖色被GB2760《食品安全国家标准 食品添加剂使用标准》和国家标准GB8817《食品添加剂焦糖色》双重管理。在GB2760《食品安全国家标准 食品添加剂使用标准》中,焦糖色可以使用于包括饮料在内的十几种食品中。 2、因工艺的不同,在焦糖色生产过程中可能会产生微量的4-MEI。该成分是在焦糖等食品的加热、烘烤和烹饪过程中形成的。在世界范围内,含有焦糖色的众多食品和饮料中都可能有微量的4-MEI。 3、国际公共健康组织,如欧洲食品安全委员会(EFSA)、加拿大卫生部等,已经证实焦糖色以及包含的微量4-MEI,对包括可乐在内的饮料和食品是安全的。 4、中国饮料工业协会相信饮料企业在严格执行GB2760《食品安全国家标准 食品添加剂使用标准》和GB8817《食品添加剂 焦糖色》、确保饮料质量合规及消费安全的同时,将积极引进国内外的先进技术、提高技术水平。 中国饮料工业协会 2013年8月9日看来焦糖色素中含有4-MEI,是肯定的,但是很微量。一些本来无毒的物质,在加热,烘烤等处理后,也可能会产生对人体有害的物质。这个应该是无法避免的。
请问谁有甲磺酸的检测方法,最好是简单的点的,易操作的
有谁做过1-丁基-3-甲基咪唑氯盐的分析?(1)色谱柱为 ODS 型C18色谱柱;流动相为水和甲醇,流动相比为水∶甲醇 = 1∶9,流速为1.0 mL/min;室温条件下检测,紫外检测波长为215 nm。(2) BDS型C18色谱柱;流动相为水(pH = 1.63)∶甲醇 = 85∶15,流速为1.0 mL/min;室温下检测,紫外检测波长为215 nm。我用第一种方法做过,在2.5分钟有一个很大的峰出来,但是接着就有一个很小的峰出来.两个峰分不开.
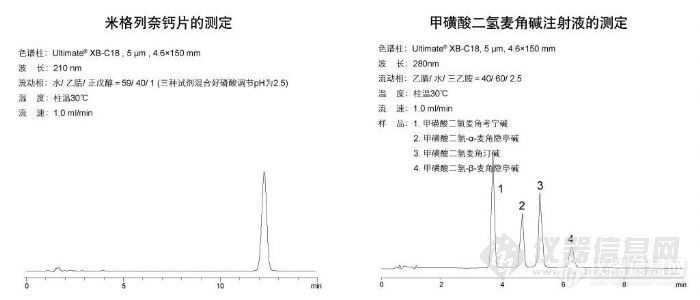
米格列奈钙片测定和甲磺酸二氢麦角碱注射液的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021813_180160_1896702_3.jpg
请问三氟甲磺酸钠、三氟甲磺酸镨如何检测含量,急需
http://www.3158.com/upfiles4/2010/08/24/15/06/07/6599bb85.jpg请教各位版友,用液相法分析甲磺酸伊马替尼(见上式),其中有个中间体是没有与甲磺酸成盐的成分。在液相的溶液条件下(水相pH2.5),能把这两种物质分开吗?甲磺酸伊马替尼是可以水解的吧?这个中间体和甲磺酸依马替尼会生成同一种物质吗?要是这样的话,那这个中间体就没法分析了吧。谢谢各位版友!
请问三氟甲磺酸钠、三氟甲磺酸镨如何检测含量,急需

遗传毒性杂质,现在是药学研究的焦点之一。甲磺酸、苯甲磺酸等磺酸盐类物质与微量的低级醇在合成反应中生成烷基磺酸酯类,这些物质可与DNA发生烷基化反应,从而可能成为引发癌症的诱因。欧洲医药评价署、美国食品和药品管理局及国际药品注册协调会议等先后对基因毒性杂质做出限度规定。具体到我们的甲磺酸加贝酯产品,需要对其中的甲磺酸乙酯的限度进行控制。溶液制备:对照品溶液 取甲磺酸乙酯适量,精密称定,用乙腈制成每毫升含1.5μg/ml的溶液,精密移取2ml加入顶空瓶中,加入3ml水,6g碘化钠后,扎盖密封。供试品溶液 取甲磺酸加贝酯适量,精密称定,按供试品100mg与乙腈1ml的比例配制供试品溶液,溶液经超声、波膜过滤处理后,精密移取2ml加入顶空瓶中,加入3ml水,3g碘化钠后,扎盖密封。色谱条件:Agilent 7890色谱仪,顶空进样器,FID检测器;色谱柱,月旭WEL-PEG20M,30m*0.32mm*0.25μm(Cat. NO:01918-32001;Ser. NO:GC20131102);进样口温度为110℃,检测器温度为260℃,氢气流速为30ml/min,空气流速为350ml/min,进样量为1mL,分流比为0.1:1。升温程序,起始温度为40℃,维持10min,然后以20℃/min的升温速率,升温至160℃,维持1min。顶空瓶平衡温度为80℃,平衡时间为30min。结果:对照液色谱图: http://ng1.17img.cn/bbsfiles/images/2014/07/201407021418_503868_1609327_3.jpg其中,时间为1.982min的保留峰为甲磺酸乙酯的衍生物。供试液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/07/201407021538_503896_1609327_3.jpg由色谱图中可以看出,样品中未检出甲磺酸乙酯。讨论:出峰时间非常的快,但是理论塔板数、分离度、对称因子等却非常给力!既得到了良好的分离效果,又尽可能的节约了分析时间。
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。

[align=center][b]甲磺酸伊马替尼片的中试质量研究[/b][/align][align=center]王淑华,臧恒昌[/align](山东大学药学院)[b]摘要:[/b]甲磺酸伊马替尼片是一种小分子靶向抑制剂,用于治疗费城染色体阳性的慢性髓性白血病的慢性期、加速期或急变期和不能切除和/或发生转移的恶性胃肠道间质瘤的成人患者。甲磺酸伊马替尼由瑞士诺华公司2001年在美国首研上市,作为肿瘤的首个靶向治疗药物面世开创了肿瘤分子靶向治疗的新时代,目前已经在全球90多个国家获得批准,美国、欧盟和其它国家还批准甲磺酸伊马替尼片用于胃肠基质瘤患者的治疗。2005年进口到中国,中文商品名是格列卫。本文按照现行药品注册法规的要求对甲磺酸伊马替尼片的制备工艺进行研究,在小试工艺处方的基础上进行中试放大,对粉碎、混合、制粒、总混、压片、包衣的工艺参数进行研究确定,并确定中试设备,用中试产品与格列卫进行全面的质量对比试验,并进行影响因素试验考察10天的研究。开发出与原研药具有相同质量的甲磺酸伊马替尼片,实现甲磺酸伊马替尼片的可工业化生产。[b]关键词:[/b]甲磺酸伊马替尼片;开发;制备工艺;[b]1 实验材料和仪器[/b]1.1实验材料[align=center][img=,572,220]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251640111718_2529_3389662_3.png!w572x220.jpg[/img][/align]1.2实验仪器[align=center][img=,573,287]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251642146696_8860_3389662_3.png!w573x287.jpg[/img][/align][b]2方法与结果 [/b] 研究内容包括中试放大3批,批量为5000片,筛选各项工艺参数、进行影响因素考察、与原研药进行全面的质量对比,最终确定了中试规模的处方、工艺、工艺参数、设备及场所。[b]2.1 中试3批样品的制备[/b]为了充分验证处方及制备工艺的可行性,优化各项工艺参数,中试制备了三批甲磺酸伊马替尼片(批号20111205、20111228、20120104),每批5000片,三批产品处方见表3[align=center][img=,555,295]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251643270416_551_3389662_3.png!w555x295.jpg[/img][/align]制备工艺:取甲磺酸伊马替尼,用万能粉碎机粉碎,筛网目数为100目,粉碎后称取处方量,备用。粉碎过筛后的甲磺酸伊马替尼、微晶纤维素、羟丙甲纤维素、交联聚维酮、二氧化硅和硬脂酸镁分别称取处方量备用;将甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中,混合9min,搅拌转速20Hz,剪切转速30Hz;在HLSG-10型混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15HZ,剪切转速15HZ,时间5min,取出后摇摆制粒机20目筛制粒;湿颗粒置60℃热风循环干燥箱中干燥,至水分为2.5%以下时停止;干燥完的颗粒取出,用摇摆制粒机24目筛整粒;整粒后的颗粒,加入交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅,置SH-20三维混合机中混合,转速为9rpm,时间为20min,然后加入硬脂酸镁,继续混合10min,出料。取样检测中间体含量,计算理论片重;将上述总混粉用ZPW-21B型旋转压片机压片,Ф9mm圆形双凸冲模,控制平均片重为理论片重±3%,硬度50-70N;LDCS型高效包衣机,出风温度:38℃;锅体转速:5-10 rpm;喷液泵转速:5-10 rpm;雾化压力:1100mbar;直喷压力:750mbar;包衣增重2%-4%;用铝塑包装机进行泡罩包装,每板10片。泡罩板外套复合膜袋。[b]2.2 工艺参数的研究2.2.1 原料药的粉碎 [/b]甲磺酸伊马替尼为水中易溶的药物,粉碎的粒度对药物溶出的影响不大,因此,确定使用湿法制粒的常规工艺参数:即万能粉碎机粉碎,筛网为100目,备用。[b]2.2.2 混合[/b]混合采用高效湿法混合制粒机,甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中混合,搅拌转速20Hz,剪切转速30Hz,分别于3min、6min、9min和12min在不同位置取样测定甲磺酸伊马替尼的含量,计算RSD值,结果见表4,中试三批的混合参数见表5。[align=center][img=,583,304]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251644390288_1847_3389662_3.png!w583x304.jpg[/img][/align]结果显示中试样品在6min时,各个位置的含量测定结果已经没有显著差异(RSD<5%),表明这时已经混合均匀,9min和12min时,物料更加均匀(RSD<2%)。为保证工艺操作的可靠性,将中试的混合时间确定为9min。[b]2.2.3 制粒 [/b]20111205批中试样品的制粒过程:在混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15Hz,根据处方筛选的结果,加入的纯化水量应为75ml,制备时先加入50ml,然后开启制粒(剪切),转速15Hz,2min后停机观察,发现软材略干,润湿不够,又加入少许,最终纯化水加入量为65 ml,制粒3min后停机观察,发现软材能够握紧成团,轻压即散,符合要求。出料后,置20目筛摇摆制粒机中制粒,湿颗粒置60℃干燥箱中干燥,至水分为2.5%以下时停止。24目筛摇摆制粒机整粒。20111228,20120104两批样品的制备均按照上述参数执行。最终确定中试的制粒参数为:搅拌转速15HZ,剪切转速15HZ,时间5min。取出后摇摆制粒机20目筛制粒。60℃干燥。水分控制小于2.5%。24目筛整粒。[b]2.2.4 总混 [/b] 由于本品制粒后需要加入较多的粉末,包括交联聚维酮、微晶纤维素、二氧化硅和硬脂酸镁,约占片芯总重的22%,所以保证粉末和颗粒的充分混合就比较关键。结合20111205批中试样品的制备,对总混时间进行了取样验证。将整粒后的颗粒与交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅同置三维混合机中混合20min,转速为9rpm,然后加入硬脂酸镁,继续混合10min。分别于15min、20min、25min和30min在混合机中物料的不同部位取样6份,测定其中甲磺酸伊马替尼的含量,计算RSD值,结果见表6,三批中试批混合参数见表7。[align=center][img=,613,321]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251656004349_7538_3389662_3.png!w613x321.jpg[/img][/align]最终确定的中试混合工艺参数为:将整粒后的颗粒与交联聚维酮、微晶纤维素(Ⅱ)、二氧化硅同置三维混合机中混合20min,转速为9rpm,然后加入硬脂酸镁,继续混合10min。[b]2.2.5 中间体含量测定[/b]总混粉取样,测定其中伊马替尼的含量,按100mg/片计算理论片重,见表8。[align=center][img=,574,92]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251656591680_4007_3389662_3.png!w574x92.jpg[/img][/align][b]2.2.6 压片 [/b]参照原研药,采用Ф9mm浅圆冲压片。单独制备了一批3000片用量的总混粉,分别压制不同硬度范围的甲磺酸伊马替尼片各约800片,以确定合适的硬度,结果见表9~11及图1。[align=center][img=,555,545]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251657410320_9022_3389662_3.png!w555x545.jpg[/img][img=,512,293]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251657512292_1250_3389662_3.png!w512x293.jpg[/img][/align]试验结果显示GYYJ01批的溶出5min明显快于格列卫,10min和15min略快于格列卫,其脆碎度为0.5%,且有裂片和断片出现,脆碎度不合格;GYYJ02批溶出曲线与格列卫基本一致,脆碎度合格;GYYJ03批溶出曲线明显慢于格列卫,脆碎度合格。因此,确定压片硬度应控制在50-70N的范围之内。三批中试样品压片参数见表12。[align=center][img=,573,160]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251658297289_6842_3389662_3.png!w573x160.jpg[/img][/align][b]2.2.7 包衣[/b]取GYYJ02批的素片,进行包衣增重的研究。分别于不同时间取出部分片剂,使得它们具有不同的包衣增重。包衣条件为:取包衣粉,用纯化水配制成固含量为13%的液体,搅拌40分钟,备用;出风温度38℃,锅体转速5-10rpm,喷液泵速度5-10rpm,侧喷压力1100mbar,直喷压力750mbar。结果见表13~14及图2[align=center][img=,596,329]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251659214722_6185_3389662_3.png!w596x329.jpg[/img][img=,532,316]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251659216782_8146_3389662_3.png!w532x316.jpg[/img][/align]试验结果显示3种不同的包衣增重对溶出曲线基本无影响,因此,确定包衣增重的范围为2%~4%。中试3批包衣结果见表15[align=center][img=,593,227]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251700491390_8973_3389662_3.png!w593x227.jpg[/img][/align][b]2.2.8 包装 [/b]包衣片用铝塑包装机包装,成形温度118℃,热封温度120℃。2.2.9 中试研究工艺参数汇总[align=center][img=,533,471]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251701345335_8652_3389662_3.png!w533x471.jpg[/img][/align][b]2.3 三批中试产品数据及与原研药的对比研究[/b]结果见表17~18及图3。[align=center][img=,651,279]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251703255806_9275_3389662_3.png!w651x279.jpg[/img][img=,605,691]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251703354406_5321_3389662_3.png!w605x691.jpg[/img][/align][align=center][img=,554,639]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251705217673_2956_3389662_3.png!w554x639.jpg[/img][img=,537,641]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251705282186_7479_3389662_3.png!w537x641.jpg[/img][/align]试验结果显示20111205、 20111228两批自研产品与原研产品格列卫,在0.1M盐酸、pH6.8磷酸盐缓冲液和pH4.5醋酸盐缓冲液和水等4种溶出介质中15分钟溶出度均超过85%,判定为体外溶出行为一致。三批中试产品的各项质量指标与格列卫一致。[b]2.4 影响因素试验[/b]取20111205批中试样品,置强光照射(照度4500Lx)、高温(60℃)、高湿(RH92.5%和RH75%)条件下各放置10天,分别于0、5、10天检测吸湿增重、性状、溶出度、有关物质、含量等各项指标。同时取对照药(格列卫,100mg),置上述条件下,于0天和10天检查相应的项目,作为对比研究。影响因素试验结果表19。[align=center][img=,565,439]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251706500796_5767_3389662_3.png!w565x439.jpg[/img][/align][align=center][/align]试验结果显示自研产品和进口原研产品在高湿RH75%±5%条件下考察10天,吸湿增重均超过5%,提示产品应注意防潮。自研产品和进口原研产品在其它3个条件下各项指标均保持稳定,无显著变化。[b]2.5 中试研究试验结果[/b]2.5.1 处方(按5000片计),见表20[align=center][img=,567,322]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251708221656_8624_3389662_3.png!w567x322.jpg[/img][/align][b]2.5.2 制备工艺 [/b](1)原辅料的处理取甲磺酸伊马替尼,用万能粉碎机粉碎,筛网目数为100目,粉碎后称取处方量,备用。粉碎过筛后的甲磺酸伊马替尼、微晶纤维素、羟丙甲纤维素、交联聚维酮、二氧化硅和硬脂酸镁分别称取处方量备用。(2)混合将甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中,混合9 min,搅拌转速20Hz,剪切转速30Hz。(3)制粒在HLSG-10型混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15HZ,剪切转速15HZ,时间5min,取出后摇摆制粒机20目筛制粒。(4)干燥湿颗粒置60℃热风循环干燥箱中干燥,至水分为2.5%以下时停止。(5)整粒干燥完的颗粒取出,用摇摆制粒机24目筛整粒。(6)总混整粒后的颗粒,加入交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅,置SH-20三维混合机中混合,转速为9rpm,时间为20min,然后加入硬脂酸镁,继续混合10min,出料。取样检测中间体含量,计算理论片重。(7)压片将上述总混粉用ZPW-21B型旋转压片机压片,Ф9mm圆形双凸冲模,控制平均片重为理论片重±3%,硬度50-70N。(8)包衣 LDCS型高效包衣机,出风温度:38℃;锅体转速:5-10 rpm;喷液泵转速:5-10 rpm;雾化压力:1100mbar;直喷压力:750 mbar;包衣增重2%-4%。(9)包装用铝塑包装机进行泡罩包装,每板10片。泡罩板外套复合膜袋。[b]2.5.3 中试研究场地[/b]固体制剂中试车间[b]2.5.4 中试设备[/b]见表2。[b]2.5.5 质量评价[/b]与原研药格列卫对比研究结果显示,中试产品的各项质量指标与格列卫相当,高温、光照、高湿三种剧烈条件下考察10天后,中试产品的各项质量指标仍与列卫相当,说明自研中试产品与原研产品质量一致。[b]3 结论[/b]因为本品原料是水溶性原料,粒度对溶出度影响不大,所以对原料前处理采用了常规机械粉碎法,过100目筛。物料混合6-12分钟都可以混匀,选择了中间点9分钟作为混合时间。根据实际情况,粘合剂水的用量由小试的2.5g/200片降到了65g/5000片。多批样品颗粒水分都小于2.5%,说明控制2.5%以下的颗粒水分适合本工艺。三批中试结果显示总混30分钟可以保证物料混合均匀。通过溶出曲线和脆碎度两个指标,考察了30-50N、50-70N、70-100N三个硬度范围,结果显示压片硬度范围在50-70N更为合理。包衣环节,考察了包衣增重2.1%、3.2%、4.2%三个梯度,对溶出曲线均无影响,最后确定包衣增重范围是2-4%。本文对甲磺酸伊马替尼片的制备工艺进行研究,用中试产品与格列卫进行全面的质量对比试验,并进行了影响因素试验考察研究,拟开发出与原研药具有相同质量的甲磺酸伊马替尼片,实现甲磺酸伊马替尼片的可工业化生产。







 我要推广仪器
我要推广仪器
 下载APP
下载APP