离子色谱分析氨基糖苷类药物及在各国药典中的应用
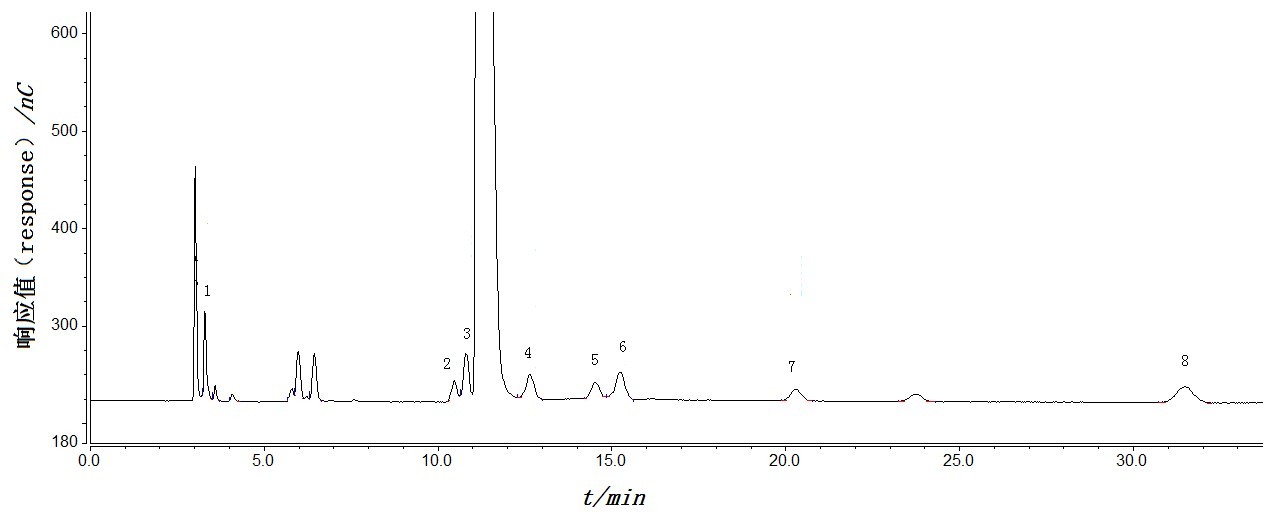
离子色谱自上世纪70年代开始经过近40多年的发展,已成为色谱分析领域中十分重要的分支,被广泛应用于无机阴阳离子、有机酸、糖醇类化合物、氨基酸、氨基糖苷类抗生素等,具有方便快速、灵敏度高、选择性好、可同时分析多种化合物、样品用量少等优点。离子色谱的检测器主要有电化学检测器与光学检测器,在药品控制领域,应用得最多的为电化学检测器,包括电导检测器和安培检测器。电导检测器主要用于测定无机阴阳离子与部分极性有机物如羧酸等。安培检测器又可分为直流安培检测器与积分安培(包括脉冲安培)检测器,其中积分安培检测器主要用于测定糖类、氨基酸类及氨基糖苷类抗生素等。氨基糖苷类抗生素具有相似的化学结构与理化性质,都是以碱性环己多元醇为苷元,与氨基糖缩合成苷,是临床应用较早的一类抗生素。氨基糖苷类抗生素根据其来源可分为发酵与半合成2种,其中发酵来源的主要有链霉素、新霉素、卡那霉素、巴龙霉素、妥布霉素、庆大霉素、核糖霉素及大观霉素等;半合成是以发酵来源的抗生素为前体,再进行结构改造而得到,主要有阿米卡星、奈替米星、异帕米星及我国自主研发的依替米星等,具有更强的抗菌活性、低耐药性及低毒性等。氨基糖苷类抗生素结构中无紫外吸收基团,难以采用常规的高效液相色谱-紫外检测器控制质量,目前国内常用的分析方法为高效液相色谱-蒸发光散射检测法(HPLC-ELSD)。由于其结构中含有多个氨基(-NH2)与羟基(-OH),在强碱性溶液中易解离成阴离子,在一定电压下,可在金电极表面发生氧化反应,实现脉冲安培检测,因此国外药典中多采用离子色谱法检测该类药物。本文概述了本实验室近十几年来采用离子色谱法分析氨基糖苷类抗生素的实例,并简述离子色谱法在各国药典中控制该类药物的应用与发展趋势。1. 硫酸阿米卡星、硫酸阿米卡星注射液与注射用硫酸阿米卡星有关物质1.1 色谱条件YMC ODS-Aq C18(4.6mm×250mm, 5µm)色谱柱,流动相为1L无二氧化碳的去离子水中加三氟乙酸20mL,五氟丙酸300μL,七氟丁酸300μL,50%(V/V)氢氧化钠溶液8mL,用50%(V/V)氢氧化钠溶液调节pH为3.3,加乙腈10mL;流速1.0 mLmin-1;柱后加碱2.1%(V/V)氢氧化钠溶液,流速为0.3mLmin-1;脉冲安培电化学检测器,工作电极为金电极(直径3mm),参比电极为Ag-AgCl复合电极,四波形检测电位(T1: 0.00~0.40s,E1: 0.1V;T2: 0.41~0.42s,E2: -2.0V;T3: 0.43s,E3: 0.6V;T4: 0.44~0.50s,E4: -0.1V)。柱温为35℃,进样量20μL。1.2 结果硫酸阿米卡星与其杂质A、杂质B、杂质 C、杂质D、杂质E、杂质G、杂质H、杂质I均能分离,见图1。阿米卡星质量浓度在0.4985~9.969 µgmL-1范围内峰面积线性关系良好,阿米卡星峰检测限为2.0ng,定量限为5.0ng。供试品溶液中除辅料峰外,各杂质均以主成分自身对照法计算,其中杂质B校正因子为1.4,杂质C校正因子为1.3,杂质D校正因子为0.8,杂质E校正因子为1.2,杂质H校正因子为1.4,杂质I校正因子为0.6。结果8批次硫酸阿米卡星原料总杂质含量为1.2%~1.7%,77批次硫酸阿米卡星注射液总杂质含量为1.1%~2.3%,10批次注射用硫酸阿米卡星总杂质含量为1.2%~2.2%。1. 杂质I 2.杂质B 3.杂质G 4.杂质A 5.杂质C 6.杂质D 7.杂质E 8.杂质H图1 硫酸阿米卡星系统适用性色谱图中国药典2020年版(ChP2020)采用高效液相色谱紫外末端吸收法测定硫酸阿米卡星及其制剂的有关物质。英国药典2024年版(BP2024)与欧洲药典11.0版(EP11.0)均采用离子色谱法测定,流动相体系均为辛烷磺酸钠-无水硫酸钠-四氢呋喃,其中四氢呋喃是影响该方法测定的关键因素,同样纯度不同品牌、甚至同一品牌不同批号的的四氢呋喃都会影响该方法的重复性。此外,EP 11.0 与BP2024的方法还存在运行时间太长大于100min,三电位检测对金电极损耗较大,盐浓度较大对仪器损耗大等缺点。本实验室同样采用离子色谱法,用多氟烷酸体系代替辛烷磺酸钠体系,简化了流动相的配制,缩短了分析时间为35min,用四电位取代三电位保护了工作电极,检测的杂质数量与杂质总量均多于ChP2020的紫外末端吸收法,可用于硫酸阿米卡星及其制剂的有关物质控制。2. 硫酸庆大霉素注射液、硫酸庆大霉素片与硫酸庆大霉素颗粒2.1 色谱条件TSK-gel ODS-81Ts C18(4.6mm×250mm,5µm)色谱柱;流动相为0.7%三氟乙酸(含0.025%五氟丙酸,50%(V/V)氢氧化钠4ml,用50%(V/V)氢氧化钠调节pH值至2.6)-乙腈(97:3);流速为1.0mLmin-1;柱后加碱为2%(V/V)氢氧化钠溶液,流速为0.3mLmin-1;脉冲安培电化学检测器,工作电极为金电极(3mm),参比电极为Ag-AgCl复合电极,四电位检测:同前;柱温为35℃;进样量20µL。2.2 结果硫酸庆大霉素含有4个主组分,分别为C1、C1a、C2a、C2,还含有结构相似的小组分西索米星与小诺霉素。该方法可完全分离4个主组分,并可同时分离出22个有关物质。庆大霉素C1a、西索米星与小诺霉组分的检测限分别为5.3ng、3.5ng与8.0ng,定量限分别为17.8ng、11.6ng与26.7ng。ChP2020采用HPLC-ELSD法测定硫酸庆大霉素注射液的组分,而BP2024与EP11.0均采用离子色谱法测定硫酸庆大霉素原料的组分与有关物质,USP现行版采用离子色谱法测定其原料的组分,均未采用离子色谱法对硫酸庆大霉素注射液进行控制。本实验室对比了离子色谱法与HPLC-ELSD法同时测定硫酸庆大霉素注射液的有关物质,发现两种方法的分离效能相当,但采用离子色谱法时各组分的响应值随其电化学活性不同而差异明显,如西索米星的响应因子大于小诺霉素,在以西索米星为外标法进行有关物质测定时,结果小于HPLC-ELSD。 3 硫酸庆大霉素片组分与有关物质3.1 色谱条件Thermo AcclaimTMAmG C18(4.6mm×150mm, 3µm)色谱柱,流动相为0.7%三氟乙酸(含0.025%五氟丙酸,50%(V/V)氢氧化钠4mL,用50%(V/V)氢氧化钠溶液调节pH至2.6)-乙腈(96.5:3.5),流速1.0mLmin-1,柱后溶液为2%(V/V)的氢氧化钠溶液,柱后加碱为0.3mLmin-1;脉冲安培电化学检测器,工作电极为金电极(直径3mm),参比电极为Ag-AgCl复合电极,四波形检测电位(T1: 0.00~0.40s,E1: 0.1V;T2: 0.41~0.42s,E2: -2.0V;T3: 0.43s,E3: 0.6V;T4: 0.44~0.50s,E4: -0.1V)。柱温为35℃,进样量20μL。3.2 结果该方法中庆大霉素C1、C1a、C2a、C2分别在1.328~132.8µgmL-1、1.606~160.6µgmL-1、7.378~737.8µgmL-1、1.276~127.6µgmL-1浓度范围内线性关系良好,回收率为98.2%~101.8%。有关物质测定中,西索米星在2.632~52.64µgmL-1、小诺霉素在2.006~25.07µgmL-1浓度范围内线性关系良好,西索米星检测限为0.01µg,小诺霉素检测限为0.02µg,各杂质与庆大霉素各组分均能完全分离,见图2。156批次中148批次的硫酸庆大霉素片各C组分的绝对含量分别为C1a为26.3%~37.1%,C2+ C2a为41.8%~49.3%,C1为16.5%~22.2%,4个组分总含量为90.6%~105.0%。148批次的有关物质为小诺霉素1.8%~2.8%,西索米星为未检出~1.5%,其他最大单杂为 0.3%~0.9%,其他总杂为1.2%~4.2%。发现其余8批次样品组分与有关物质均不符合规定,原因为企业采用不符合标准规定的原料所致。1-5,7-8.未知杂质 6. 西索米星 9.小诺霉素图2 硫酸庆大霉素片有关物质典型色谱图ChP2020采用微生物检定法控制其含量,未控制有关物质。BP2024、EP11.0与USP现行版均未收载该品种。本实验室在参考国外药典离子色谱法测定其原料的基础上建立了硫酸庆大霉素片组分与有关物质的方法。方法对乙腈的比例进行了调整,工作电位由四电位取代三电位,可有效的分离硫酸庆大霉素片各组分与各杂质。4.硫酸庆大霉素颗粒组分与有关物质 4.1 色谱条件YMC-Pack Pro C18 RS(4.6×250mm,5μm)色谱柱,流动相为1.6%三氟乙酸(含0.05%五氟丙酸,50%(V/V)氢氧化钠8ml,用50%(V/V)氢氧化钠溶液调节pH值至2.6)-乙腈(94:6),流速1.0 mLmin-1,柱后加碱为2%(V/V)的氢氧化钠溶液,柱后加碱为0.3mLmin-1;脉冲安培电化学检测器,工作电极为金电极(直径3mm),参比电极为Ag-AgCl复合电极,四波形检测电位(T1: 0.00~0.40s,E1: 0.1V;T2: 0.41~0.42s,E2: -2.0V;T3: 0.43s,E3: 0.6V;T4: 0.44~0.50s,E4: -0.1V)。柱温为35℃,进样量20μL。4.2 结果硫酸庆大霉素颗粒的辅料主要为蔗糖,含量较高,与主成分的比例约为200:1,出峰时间约为5min。采用硫酸庆大霉素片的方法测定颗粒时,蔗糖的拖尾峰会导致前15min的基线抬高,严重干扰颗粒有关物质的测定。因此本实验室在硫酸庆大霉素方法的基础上增加了三氟乙酸、五氟丙酸与乙腈的比例,成功解决了蔗糖对硫酸庆大霉素颗粒有关物质测定的干扰。该方法中庆大霉素C1、C1a、C2a、C2分别在5.264~131.6µgmL-1、5.032~125.8µgmL-1、5.595~139.9µgmL-1、3.410~85.24µgmL-1浓度范围内线性关系良好,回收率为98.7%~100.8%。有关物质测定中,西索米星在1.987~39.74µgmL-1、小诺霉素在2.045~51.13µgmL-1浓度范围内线性关系良好,西索米星检测限为0.003µg,小诺霉素检测限为0.01µg,各杂质与庆大霉素各组分均能完全分离,见图3。1-14,16-18-未知杂质;15-西索米星;19-小诺霉素图3 硫酸庆大霉素颗粒有关物质典型色谱图5.盐酸大观霉素与注射用盐酸大观霉素有关物质 5.1 色谱条件采用离子色谱法及HPLC-ELSD法同时分析注射用盐酸大观霉素的有关物质。两法色谱柱均为Apollo C18 (250mm× 4.6mm,5µm),流动相均为0.1molL-1三氟乙酸溶液,柱温均为30℃,进样量均为20µL。离子色谱检测:柱后加减为21g/L氢氧化钠溶液,流速0.5mlmin-1,工作电极为金电极(直径3mm),参比电极为Ag-AgCl复合电极,四波形检测电位(T1: 0.00~0.40s,E1: 0.1V;T2: 0.41~0.42s,E2: -2.0V;T3: 0.43s,E3: 0.6V;T4: 0.44~0.50s,E4: -0.1V)。ELSD检测:漂移管温度110℃,载气流速2.6Lmin-1,增益1。5.2 结果ChP2020采用HPLC-ELSD法控制其原料,BP2024与EP11.0采用离子色谱法控制其原料。注射用盐酸大观霉素为无菌原料直接分装,本实验室参考国外药典方法测定了盐酸大观霉素及其制剂的有关物质,并同时与HPLC-ELSD方法进行比较。结果两种方法检测出的有关物质种类和数量基本一致,但离子色谱灵敏度比ELSD高,离子色谱检测限为2.4ng,ELSD为72.8ng。两种方法测定的31批次注射用盐酸大观霉素,杂质D与杂质E结果基本一致,但杂质A、4R-双氢大观霉素及总杂质结果差异较大,原因为杂质A、4R-双氢大观霉素杂质在两种检测器上响应不一致。因此采用离子色谱测定时需对杂质A与4R-双氢大观霉素杂质进行校正因子计算,按校正因子计算后的有关物质结果两种方法基本一致。6.青霉胺与青霉胺片含量与有关物质6.1 色谱条件Dikma Spursil C18(4.6mm×250mm,5µm)色谱柱;流动相为5.3g无水磷酸二氢钠-0.25g己烷磺酸钠,加去离子水1L溶解后,用磷酸调节pH值为2.85,加乙腈9ml;流速为1.0mLmin-1;柱后加碱为21gL-1氢氧化钠溶液,流速为0.3mLmin-1;脉冲积分安培电化学检测器,工作电极为金电极(1mm),参比电极为Ag-AgCl复合电极,六电位检测(T1为0~0.04s,E1为0.13V;T2为0.05~0.21s,E2为0.33V;T3为0.22~0.46s,E3为0.55V;T4为0.47~0.56s,E4为0.33V;T5为0.57~0.58s,E5为-2.0V;T6为0.59~0.60s,E6为0.93~0.13V);柱温为30℃;进样量20µL。6.2 结果含量测定方面,青霉胺浓度在49.88~199.5µgmL-1范围内线性关系良好,回收率为98.4%~101.5%,31批次青霉胺片含量为97.6%~101.5%。有关物质测定方面,各杂质与主成分青霉胺均能完全分离(见图4),青霉胺浓度在3.118~49.88µgmL-1,青霉胺二硫化物杂质浓度在1.616~19.39µgmL-1范围内线性关系均良好,青霉胺与青霉胺二硫化物杂质的检测限均为0.02µg;青霉胺二硫化物结果为0.4%~0.8%,最大单杂为0.9%~2.9%,其他总杂为2.4%~7.3%。1. EDTA 2.辅料3~8.未知杂质 9.青霉胺10.青霉胺二硫化物图5 青霉胺片有关物质典型色谱图ChP2020采用电位滴定法测定其含量,USP现行版采用HPLC法测定其含量,二者均未控制其有关物质。青霉胺虽不属于氨基糖苷类抗生素,但其结构中含有多个氨基与羧基,无共轭双键,同样可以采用离子色谱法测定。离子色谱法测定该品种的关键点为检测电位的选择,直接采用糖四电位时主成分响应很弱,采用仪器自带的六电位时峰型严重拖尾,因此本实验室采用循环伏安法分别对青霉胺与杂质青霉胺二硫化物进行扫描,确定了最佳的六电位波形,解决了主成分严重拖尾的问题。讨论讨论1: 操作过程中遇到的问题与解决方法离子色谱电化学检测在操作过程中常存在背景信号较高、基线噪音较大,重复性差等问题,导致试验耗时耗力,进展缓慢。如硫酸阿米卡星及其制剂测定过程中会出现响应信号下降的现象,原因为流动相中的三氟乙酸可使金电极表面钝化,使用一段时间后需用水擦拭金电极。硫酸庆大霉素制剂测定过程中,出现了背景信号缓慢增加,基线噪音增大的情况,使用一段时间后需用硝酸冲洗管路或打磨电极。为解决该问题,本实验室与离子色谱工程师们查找问题与原因,耗时近3年,终于初步解决了上述问题。首先,所有涉及的容器、试剂与过滤装置均应单独使用,试剂均应为高纯度试剂。其次,对仪器的部分管路用聚醚醚酮材料的管线取代原白色塑料管线,降低管路的透氧性。再次,仪器使用前分别用1.5molL-1的硝酸溶液、2.4gL-1的EDTA溶液、乙腈与去离子水依次冲洗管路。接着,使用时分别对流动相、柱后碱液的水离线脱气15min,除去溶解在其中的氧气,脱气完成后再用氮气或氦气保护。使用时所有的管路须充满液体,防止氧气进入系统中导致重复性降低。最后,更换了进样阀。初步解决了重复性差的问题,但测定时仍需要在碱液中加入一定浓度的EDTA,降低金属离子的影响。虽然重复性差的问题初步得到解决,但背景信号较高,剂型噪音较大等问题在日常操作中还存在着,还需要继续磨合。讨论2:各国药典中离子色谱法分析氨基糖苷类药物的情况(1)中国药典ChP2005年版在“附录V D 高效液相色谱法”检测器下提到了电化学检测器。从2010年版开始在附录中单独列出了“离子色谱法”,对离子色谱的色谱柱、洗脱液、检测器、测定法均进行了详细说明。直到2015年版才首次将该法收录至正文中,涉及的品种为硫酸依替米星,检测项目为有关物质与含量,同时还设有第二法为HPLC-ELSD法,二者选其一。现行2020年版药典仍沿用2015年版方法测定硫酸依替米星。收载的氨基糖苷类药物主要都采用HPLC-ELSD法。硫酸依替米星是我国自主研发的一种半合成氨基糖苷类抗菌药物,也是ChP 2020年版唯一一个采用离子色谱法安培检测器控制的品种。有关物质方法与含量测定方法均一致,为采用C18色谱柱,以0.2molL-1三氟醋酸溶液[含0.05%五氟丙酸、1.5gL-1无水硫酸钠、0.8%(V/V)的50%氢氧化钠溶液、用50%氢氧化钠溶液调节pH值至3.5]-乙腈(96:4)为流动相,四电位检测,柱后加碱(50%氢氧化钠溶液1→25),柱后流速为0.5mLmin-1。(2)国外药典美国药典USP25-NF20首次采用高容量的三乙胺阴离子交换色谱柱,以氢氧化钠为淋洗液测定了阿米卡星(包括硫酸阿米卡星及阿米卡星注射液)、卡那霉素(包括硫酸卡那霉素、卡那霉素注射液及硫酸卡那霉素胶囊)的含量。随后,USP27-NF22开始采用耐强酸、强碱和高浓度盐的聚苯乙烯-二乙烯基苯共聚物填料色谱柱代替传统的阴离子交换柱,并首次用四电位取代三电位测定了硫酸链霉素原料、硫酸链霉素注射液及注射用硫酸链霉素的含量。随着离子色谱不断发展,USP37-NF32及之后的版本用十八烷基键合硅胶代替了聚苯乙烯-二乙烯基苯共聚物色谱柱,流动相以烷基化有机酸如三氟乙酸、五氟丙酸等作为离子对试剂测定庆大霉素原料的组分。该方法采用柱后加碱的模式,较美国药典常用的氢氧化钠淋洗液体系更能避免空气中二氧化碳的影响,分析系统更稳定。BP从2002年版、EP从4.0版开始收载了硫酸新霉素的离子色谱方法,方法采用柱后加减模式测定了硫酸新霉素原料的有关物质。随后,BP2003年版、EP5.0版及之后的版本陆续将离子色谱法应用于奈替米星、妥布霉素、庆大霉素、大观霉素及阿米卡星等品种。方法的共同特点为采用耐强酸碱的聚苯乙烯-二乙烯基苯柱或耐酸的C18柱,以烷基磺酸盐或三氟乙酸等离子对试剂作为流动相,与氨基糖苷类药物形成离子对增强其保留,再加入少量的有机改进剂改善分离,三电位检测。直到BP2007年版、EP6.0版开始陆续采用更为普及的辛烷基键合硅胶或十八烷基键合硅胶色谱柱测定了盐酸大观霉素、硫酸庆大霉素、阿米卡星与硫酸阿米卡星等。其中从BP2011年版、EP7.0版开始,硫酸庆大霉素有关物质与组分方法中,流动相由烷基磺酸盐体系变更为三氟乙酸-五氟丙酸体系,减少了流动相中的盐在金电极表面沉积并使检测信号更稳定。发展趋势与展望中国药典是药品研制、生产、经营、使用和监督管理等均应遵循的法定依据,是我国保证药品质量的法典。中国药典具有使用范围广,权威性强的特点,因此其收载的质量标准应具有操作性强、重现性好、耐用性好、成本适中等特点。目前中国药典中采用离子色谱安培检测法测定的品种仅硫酸依替米星一个,而国外药典多采用安培检测法测定氨基糖苷类药物。离子色谱安培检测法在中国药典中发展缓慢的原因主要有2点:一是国内外离子色谱仪的普及率不同。国内制药企业规模参差不齐,离子色谱仪价格较高,仅一些规模较大的企业采购了离子色谱仪;而国外制药企业规模通常较大,大多有条件购买价格昂贵的仪器。二是国内外离子色谱仪使用情况不同。国内使用离子色谱电导检测比较多,而国外电导检测与安培检测发展基本持平。由于离子色谱安培检测器在分析无紫外吸收或紫外吸收较弱的药物方面具有一定的优势,无需衍生化可直接检测,灵敏度高、选择性好,具有一定的发展前景。而且目前国产离子色谱仪蓬勃发展,日趋成熟与稳定,为今后离子色谱在药物分析方面提供了更多的技术支持和选择性。但相关离子色谱生产企业也需解决操作过程中仪器存在的一些问题,如提高仪器的重复性和易操作性,使离子色谱在今后的应用更加深入和广泛。本文作者:李茜,王立萍,刘英*(河南省药品医疗器械检验院,郑州,450018)作者简介:李茜,女,副主任药师 研究方向:抗生素质量分析与质量控制*通讯作者:刘英,女,主任药师 研究方向:抗生素质量分析与质量控制


























 我要推广仪器
我要推广仪器
 下载APP
下载APP