高分子表征技术专题——二维相关红外光谱分析技术在高分子表征中的应用
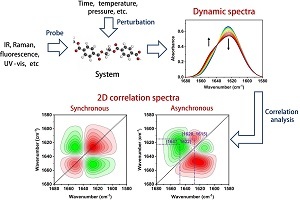
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。更多专题内容详见:高分子表征技术专题 高分子表征技术专题前言孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!二维相关红外光谱分析技术在高分子表征中的应用Applications of Two-dimensional Correlation Infrared Spectroscopy in the Characterization of Polymers本文作者:侯磊,武培怡 作者机构:东华大学化学化工与生物工程学院,上海,201620作者简介:武培怡,男,1968年生. 1985年,南京大学化学系获学士学位,1998年,德国ESSEN大学获博士学位. 1998~2000年在日本触媒研究中心从事研究工作,2000~2017年任复旦大学高分子科学系教授,2017年起任东华大学化学化工与生物工程学院教授. 2001年入选上海市科委启明星计划、上海市教委曙光计划,2003年入选上海市科委白玉兰科技人才计划,2004年入选上海市科委启明星跟踪计划,获得国家杰出青年基金资助、上海市引进海外高层次留学人员专项资金资助,2005年度入选教育部首届新世纪人才计划,2007年入选上海市优秀学科带头人计划,2016年入选英国皇家化学会会士,2017年获陶氏化学“Dow Innovation Challenge Award”. 主要研究方向包括二维相关光谱在聚合物体系中的应用、智能仿生材料、聚合物功能膜等.摘要二维相关光谱作为一种先进的光谱分析方法,具有提高谱图分辨率、解析动态过程等优势,近来在高分子表征中引起了越来越多的关注. 高分子体系涉及了丰富的相互作用和复杂的结构,分子光谱是常用的表征手段,而借助二维相关光谱分析技术,能够有效识别精细结构、判别动态变化机制,从而显著丰富和完善分析结果. 本文重点围绕二维相关红外光谱,简述了发展历史和基本原理,随后结合实际过程,介绍了相关实验和分析技巧,最后列举了其在高分子表征中的典型应用,展示了二维相关红外光谱分析的特点,具体涉及温度响应高分子的响应机制、可拉伸离子导体中复杂相互作用、小分子在聚合物基质中的扩散、天然高分子的结构表征等研究. 希望通过本文的介绍,能够帮助读者更好地理解二维相关光谱,进一步拓展其在高分子领域中的应用.AbstractTwo-dimensional correlation spectroscopy (2Dcos) is an advanced analysis method, which holds great advantages in improving spectral resolutions and interpreting dynamic processes, and has attracted great attention in the field of polymers. Molecular spectroscopy is frequently applied in the characterization of polymers, which involves abundant molecular interactions and complex structures. Under the help of 2Dcos analysis, fine structures as well as dynamic mechanisms within the polymer systems can be effectively identified, thus significantly enriching and improving the analysis results. In this paper, we will mainly focus on the two-dimensional correlation infrared spectroscopy (2DIR). Firstly, the history and basic principles of 2Dcos are briefly introduced. Then, some relevant experimental and analytical techniques are presented based on the actual process. Finally, typical applications of 2DIR in the polymer characterization are demonstrated and the features thereinto are also shown. Particularly, the response mechanisms of temperature-responsive polymers, complex molecular interactions in stretchable ionic conductors, diffusion processes of small molecules in polymer matrix and structures of natural polymers are investigated. It is hoped that this paper will help readers better understand 2Dcos and further expand its applications in the field of polymers.关键词分子光谱 二维相关光谱 高分子 分子相互作用 KeywordsMolecular spectroscopy Two-dimensional correlation spectroscopy Polymer Molecular interactions 高分子材料体系涉及丰富的相互作用和多级结构,这是决定材料最终性能的关键. 分子光谱(红外、拉曼光谱)作为表征高分子材料的常用手段,一方面可以检测不同化学结构/组分所对应的官能团,依据特征吸收峰强度和位置,实现对高分子化学结构的鉴别,另一方面,可以基于不同官能团特征吸收峰的强度和位置变化,判别基团所处的物理或化学环境,实现对体系中复杂相互作用的解析. 随着高分子材料的发展,体系趋向多样化、多功能化,而传统的一维分子光谱存在谱峰重叠严重、分辨能力有限等问题,一定程度限制了分子光谱在复杂高分子体系的应用拓展.二维相关光谱(Two-dimensional correlation spectroscopy,2Dcos)作为一种先进的光谱分析手段,尤其适合于从分子水平探讨各类外扰作用下复杂高分子体系涉及的结构和相互作用变化. 相较于传统的一维光谱,二维相关光谱的优势在于:(1)对于包含许多重叠峰的复杂谱图,起到图谱简化的作用;(2)通过将原始谱图在第二维度上延伸,能够明显提高原始一维谱图的分辨率;(3)谱峰的相关性可帮助判断体系中的相互作用以及峰归属;(4)可用于确定外界刺激下不同过程的发生次序. 本文首先将结合二维相关光谱的发展历史,介绍其基本原理. 其次,围绕动态谱图获取和二维相关分析,介绍二维相关光谱的一些实验和分析技巧. 最后,结合具体体系,重点阐述二维相关光谱在高分子表征中的应用.1 基本原理1.1 发展历史二维相关光谱分析方法的基本概念最早起源于核磁共振(NMR)领域. 二维核磁共振(2DNMR)谱通过多脉冲技术激发核自旋,采集原子核自旋弛豫过程的衰减信号,最后经双重傅里叶变换得到[1]. 通过将核磁信号扩展到第二维度,可以显著提高谱图的分辨率,并且有效简化包含许多重叠峰的复杂光谱. 与此同时,通过选择相关的光谱信号,可以鉴别和研究分子内/间的相互作用. 尽管二维光谱技术在核磁领域取得了快速发展,却在很长一段时间内未能深入到其他光谱分支,如红外、拉曼、紫外-可见吸收、荧光光谱等. 阻碍二维光谱技术发展的一个根本原因在于多重射频脉冲的二维核磁技术可以成功地在精密而昂贵的核磁仪器上实施,却不能在普通的红外、拉曼和紫外-可见吸收等光谱仪器上实现. 因为这类光谱的时间标尺(time scale)远小于核磁共振[2]. 一般来说,核磁时间标尺数量级在毫秒到微秒之间,而红外吸收光谱观察分子振动的时间标尺在皮秒数量级,因此产生二维红外光谱必须采用特殊的新途径.二维相关光谱概念上的突破是由特拉华大学(University of Delaware)的化学家Noda[3,4]提出的. 他把核磁实验中的多重射频励磁看作是一种对体系的外扰(外部扰动). 施加于体系的外扰可以多种多样,如热、磁、机械、电场、化学甚至声波等. 每种外扰对体系的影响是独特而有选择性的,并由特定的宏观刺激和分子相互作用的机理所决定. 因此,包含在动态光谱中的信息类型是由外扰的方式和电磁波的种类所决定的. 外扰的波形没有任何限制,从简单的正弦波、脉冲、到随机的噪音或静态的物理量(如时间、温度、压力等)的变化均可应用于外扰. 由此,Noda设计出一种完全不同的二维光谱实验技术,他用外扰来激发被检测体系的分子,由于被激发分子的弛豫过程慢于振动光谱的时间标尺,因而可使用时间或温度等外扰分辨振动光谱(红外、拉曼)技术来跟踪研究被检测体系受外界扰动而产生的动态变化,结合数学中的相关分析技术,将原有的光谱信号扩展到第二维度,从而得到二维相关光谱(如图1所示). 二维相关光谱实际研究的就是动态光谱的变化[5,6]. 此后,随着二维相关光谱技术的发展,逐渐在荧光光谱、X射线衍射谱、凝胶渗透色谱等也得到了应用. 总体而言,二维相关光谱分析在红外光谱中的应用最为成功,这主要是由于红外光谱的信噪比相对较高,具有高灵敏度、高选择性和非破坏性等特点,能够在分子结构和链段运动等方面提供丰富信息. 另一方面,红外光谱的谱峰重叠严重,解析起来存在一定困难,二维相关光谱的引入可以很好地解决这一问题. Fig. 1 Acquisition procedure of generalized 2D correlation spectra. In the 2D synchronous and asynchronous spectra, red colors represent positive intensities while green colors represent negative ones.1.2 计算原理二维相关光谱考虑外扰变量下(如时间、温度、压力、浓度、电场、磁场等)光谱强度y(v, p)的变化情况,其中v为光谱变量,可以为任何光谱量化的参数,如红外波数、拉曼位移、紫外波长、X射线散射角等,p为外扰变量,可以是任意合理的物理或化学变量,如时间、温度、压力、电场强度、浓度、pH、离子强度等. 对于体系在一定外扰区间(1~N)下引起的动态光谱y˜(v, p)定义为[2,5]:y¯(v)为体系的参考光谱,通常选为平均谱. 参考光谱的定义为实际过程中,可以选择某一个参考点p = Pref处的光谱作为参考光谱. 参考点可以是实验的初始状态或结束状态,也可以直接简单地设为0,这种情况下,动态光谱即为我们观察到的光谱强度.二维相关强度X(v1, v2)表示在外扰变量区间内,对光谱变量v1和v2光谱强度变化y˜(v, p)的函数进行比较. 由于相关函数是计算2个互不依赖的光谱变量v1和v2处强度的变化,因此可以将X(v1, v2)转变为复数形式[2]:这里,组成复数的相互垂直的实部和虚部分别称作同步和异步二维相关强度. 同步二维相关强度Ф(v1, v2)表示随着p值的变化,v1和v2处光谱强度的相似性变化,而异步二维相关强度Ѱ(v1, v2)则表示光谱强度的相异性变化.二维相关光谱的快速计算方式在于对动态光谱进行Hilbert-Noda变换,将其从外扰域转换到频率域上,最终得到二维相关光谱[2,5].二维相关同步谱:二维相关异步谱:其中Mjk代表Hilbert-Noda转变矩阵的第j行第k列的元素,表示为:1.3 解谱规则二维相关光谱图包含同步谱和异步谱2类,图1展示了典型的同步和异步谱图.1.3.1 二维相关光谱同步谱图二维相关光谱同步谱图表现了给定2波数v1和v2处光谱强度的同步或者一致变化. 同步谱图沿对角线(对应于光谱坐标v1 = v2)方向对称,其中相关峰可以出现在对角线上,也可以出现在对角线外. 落在对角线上的相关峰称作自动峰,自动峰强度对应于外扰过程中光谱变化的自相关函数. 在同步谱中,自动峰的强度始终为正,代表了对应波数下光谱强度动态波动的整体程度. 所以,在动态谱图中表现出更大程度强度变化的区域对应的自动峰越强,而那些基本保持不变的峰自动峰强度小甚至没有自动峰. 交叉峰处于同步谱图的非对角线区域,表现了不同波数光谱信号的同步变化. 这样一种同步的变化,反过来,预示着2波数间可能存在一定的相关性. 尽管自动峰的强度始终为正,但交叉峰的强度可正可负. 如果2波数的交叉峰为正,说明这2个波数对应的光谱强度在外扰下同时增加或者同时降低;如果两波数的交叉峰为负,说明这2个波数对应的光谱强度一个增加另一个降低.1.3.2 二维相关光谱异步谱图异步谱图呈现了2个给定波数v1和v2处光谱强度的异步或者相继变化,它关于对角线反对称. 异步谱图中只有交叉峰,而无自动峰. 异步交叉峰只有在2个给定波数的光谱强度发生异相(如延迟或加快)变化时才出现. 这一特点尤其可以帮助区分光谱中的来源不同的重叠峰. 于是,外扰过程中,混合物中的不同组分、材料中的不同相或者化学基团经历不同的变化对光谱强度的贡献能够得以辨别. 即使是2个谱带靠的很近,只要它们的瞬间特征或者时间依赖光谱强度变化模式存在本质不同,它们之间便会出现异步交叉峰. 所以异步交叉峰的出现意味着这些谱带有着不同的来源或者是不同分子环境下的官能团. 异步谱图的交叉峰可正可负,而异步谱图中交叉峰的符号可以用来辅助判断谱带在外扰过程中的变化次序.1.3.3 二维相关光谱读谱规则利用同步和异步谱图的交叉峰,可以获得外扰条件下光谱强度发生变化的先后次序关系. 为方便表述,将同步谱图中(v1, v2)处的峰强度记为Φ(v1, v2),将异步谱图中(v1, v2)处的峰强度记为Ψ(v1, v2). 根据Noda规则[5]:(1)当Φ(v1, v2) 0时,如果Ψ(v1, v2) 0,则v1谱带处的强度变化发生先于v2谱带处的强度变化(表示为v1→v2),而如果Ψ(v1, v2) 0,则v2→v1;(2)当Φ(v1, v2) 0时,如果Ψ(v1, v2) 0,则v2→v1,而如果Ψ(v1, v2) 0,则v1→v2. 简单说来,如果(v1, v2)在同步和异步谱图的交叉峰符号一致(都为正或者都为负),则v1→v2;如果(v1, v2)在同步和异步谱图的交叉峰符号不一致(一个为正而另一个为负),则v2→v1.2 实验技巧二维相关光谱作为一种有效的光谱分析手段,是针对一系列动态光谱的数学分析,具体可分为2个过程:动态谱图获取和二维相关分析. 本节将结合实际操作过程,介绍二维相关红外光谱的一些实验和分析技巧.2.1 动态谱图获取2.1.1 样品制备对于固体聚合物样品,溴化钾压片法制备的样品可直接用于透射红外光谱测试;另外,还可使用溶液铸膜(solution casting)法在红外窗片上直接制备得到适合透射红外光谱测试的薄膜. 对于溶液样品,主要应考虑样品的密封问题,避免测试过程中溶剂的挥发. 此外,水溶液或者水凝胶样品,为避免H2O分子的红外吸收对高分子链上C―H和C=O基团吸收峰的影响,可以用D2O作溶剂.2.1.2 测试条件测试模式方面,为得到高信噪比的红外光谱图,一般使用透射模式进行数据采集. 特殊的样品也可选用其他附件,例如对样品表面进行研究时可选用ATR附件. 测试条件方面,为兼顾扫描时间和信噪比,可设置红外谱图分辨率为4 cm-1,扫描次数为32次.2.1.3 测试环境二维相关光谱的特点在于只对光谱的变化敏感,能够显著放大一系列动态光谱的变化情况. 不论样品浓度、厚度如何,如果其处于静态,不发生变化,则对应的二维相关光谱无任何信号. 因此,为了使二维相关光谱的信号只来源于样品本身的结构变化,需要保证测试过程中环境的相对稳定,排除测试环境变化引起的水或二氧化碳吸收峰变化的干扰. 通常,可以借助干燥空气或者氮气吹扫,待测试环境稳定后进行背景采集,随后开展一系列动态光谱的采集.2.2 二维相关光谱分析将采集的一系列动态光谱在特定的软件上进行数学处理,即可得到二维相关光谱同步和异步谱图. 目前,能够快速获得二维相关光谱的软件种类很多[7],大都是免费获取或者是商业化的软件,包括2D Shige、TDCOS、Mat2DCorr、2DCS、Midas 2010、R corr2D、Python Scikit Spectra、Python NumPy等. 关于二维相关光谱的谱图分析,重点在两部分:精细结构的分辨和动态过程的解析. 二维相关光谱异步谱可以区分光谱中来源不同的重叠峰,将异步谱中谱峰对应的波数进行基团归属,即可分辨体系的精细结构. 此外,通过结合同步谱和异步谱交叉峰的符号,可以获得外扰条件下光谱强度发生变化的先后次序关系. 为了方便解析复杂体系谱峰响应的先后次序,根据Noda规则,本课题组提出了一种简便的判断方式[8]. 如表1、2所示,分别读出了图1异步谱中所有谱峰对应的波数及其在同步和异步谱中交叉峰的符号(强度正负),之后将其对应一一相乘,结果如表3所示. 该表中每一个正值都代表它所对应的横轴的波数先于或快于纵轴的波数响应,而每一个负值代表它所对应的横轴的波数后于或慢于纵轴的波数响应. 基于此,可以直观地得出对应动态过程的谱峰响应次序(“→”表示先于或快于):1647→1628→1622→1615 cm-1.Table 1 Signs of cross-peaks in synchronous spectrum (corresponding to Fig. 1).Table 2 Signs of cross-peaks in synchronous spectrum (corresponding to Fig. 1).Table 3 The final results of multiplication on the signs of each cross-peak in synchronous and asynchronous spectra.3 典型应用基于二维相关光谱在判断精细结构和解析动态过程的优势,本节将结合本课题组的研究工作,介绍二维相关光谱在高分子表征中的应用,主要涉及温度响应高分子的响应机制、可拉伸离子导体中复杂相互作用、小分子在聚合物基质中的扩散机理等.3.1 温度响应高分子的响应机制温度响应高分子能够在外界温度发生变化时改变自身的物理或化学性质,形成对环境的感应并产生反馈,在智能传感、药物缓释、可控驱动、过滤分离、智能窗户等领域得到了广泛关注和应用[9~11]. 温度响应高分子的响应过程往往源于分子结构或链构象的变化,分子光谱(红外、拉曼光谱)对分子基团及相应的相互作用十分敏感,非常适合于研究其中的响应机理. 传统的一维分子光谱存在谱峰重叠严重、分辨能力低以及难以捕捉动态过程等不足,借助二维相关光谱分析,可以对温度响应高分子的精细结构和动态响应机制进行深入解析,探讨其中的构效关系.聚(N-异丙基丙烯酰胺)(PNIPAM)在水溶液中呈现LCST (lower critical solution temperature)型转变,即升温过程发生相分离,相转变温度约为32 ℃[12]. PNIPAM分子链同时存在亲水的酰胺基团和疏水的碳链骨架、异丙基侧基,利用变温红外光谱对PNIPAM水溶液升温过程进行跟踪,观察到vas(CH3)和vs(CH2)吸收峰波数的降低以及Amide I区域1625和1649 cm-1处吸收峰的相互转化,表明聚合物链C―H基团的脱水和分子间/内氢键C=O… H―N的形成. 基于二维相关光谱分析,获取了PNIPAM水溶液相分离的微观动力学机理:温度升高首先发生侧基CH3的两步脱水,随后是主链的塌缩和聚集,最后为酰胺氢键的形成,并最终导致了相分离[13].PNIPAM的LCST型转变对溶剂组成也十分敏感. 尽管水和甲醇都是PNIPAM的良溶剂,但在两者以一定比例混合的状态下对PNIPAM则为不良溶剂. 例如:当甲醇和水的体积比为0.35:0.65时,PNIPAM在该混合溶剂中的LCST约为-7.5 ℃,这种现象称为“共不溶”现象. 利用红外光谱和二维相关光谱分析研究PNIPAM在水/甲醇混合溶剂中温度响应行为[14],传统一维红外光谱分析表明,相比于纯水溶液,PNIPAM链在水/甲醇混合溶剂中处于塌缩的状态,并且PNIPAM和甲醇的相互作用明显被削弱了,这主要归因于混合溶剂中水-甲醇团簇的形成导致了PNIPAM链水合位点的减少. 进一步的二维相关红外光谱分析证实了水-甲醇团簇对PNIPAM链水合过程的抑制作用.除此之外,本课题组还探讨了其他LCST型聚合物的转变机理[15~19]、共聚(无规共聚、嵌段共聚)结构对温敏聚合物相变行为的影响[20~22]、温度响应水/微凝胶的体积转变过程[23~25]等,相关工作已进行过系统总结[26,27],这里不再赘述.水凝胶结构与生物组织十分相近,在仿生皮肤等领域获得了广泛关注. 将两性离子单体与丙烯酸(acrylate acid, AA)共聚,通过调节盐浓度,制备得到具有优异可塑性、可拉伸性、自愈合性的超分子聚电解质水凝胶[28]. 同时,聚电解质的离子传输性质赋予了水凝胶对温度、应变、应力的多重感知功能. 基于对干态和湿态凝胶的红外光谱解析,获取了该水凝胶涉及的丰富的分子间/内相互作用,包括聚丙烯酸(PAA)链段羧基之间的氢键相互作用、两性离子链段中磺酸根与季铵盐的静电相互作用、PAA链段羧酸根和两性离子链段季铵盐的静电相互作用等,而这些丰富的分子间/内相互作用是该超分子水凝胶力学性能的决定性因素. 在此基础上,用甲基丙烯酸(methyacrylate acid, MAA)取代丙烯酸,即在PAA链段引入疏水的α-甲基,通过调节MAA和两性离子单体的比例,实现了超分子水凝胶在LCST和UCST (upper critical solution temperature)行为之间的转变[29],如图2所示. 具体地,当两性离子单体与MAA质量比大于1时,聚合物在水溶液中表现出UCST行为;当两性离子单体与MAA质量比等于1时,聚合物在宽的温度范围(10~80 ℃)内均不溶于水;两性离子单体与MAA质量比小于1时,聚合物在水溶液中表现出LCST行为. 同时,LCST和UCST可以通过两性离子和MAA单体的共聚比例方便地进行调节. 二维相关红外光谱从分子水平有效揭示了这一体系独特相行为的产生原因. 结果表明,羰基氢键结构的转化是LCST型水凝胶相行为的驱动力,而磺酸根涉及相互作用(水合作用、静电作用等)的变化是UCST型水凝胶相行为的驱动力.Fig. 2 (a) The chemical structure of the polyzwitterion Turbidity curves and typical photos for the (b) UCST- and (c) LCST-type hydrogels Temperature-dependent FTIR spectra (d, e) and 2D correlation spectra (f, g) of typical UCST- and LCST-type hydrogels (Reprinted with permission from Ref.[29] Copyright (2018) American Chemical Society).在天然的阳离子多糖(季铵化壳聚糖)中原位聚合亲水的阴离子单体(AA),构筑了具有温度、pH、机械力、电学等刺激响应行为的双网络聚电解质水凝胶. 该水凝胶同时集成了生物相容、离子传输、黏附、可拉伸、自愈合等多种功能,可作为仿生离子皮肤用于监测压力、温度、pH、电信号等刺激引起的生理信号变化[30]. 值得注意的是,该离子皮肤具有温度可调的黏附性,即升温黏附强度提升,降温黏附强度下降,例如水凝胶在猪皮上37 ℃下的黏附强度是20 ℃下的5.5倍,且具有良好的循环稳定性,这主要源于聚电解质水凝胶的UCST型转变. 季铵化壳聚糖由疏水主链和亲水的季铵盐基团组成,具有两亲性结构,通过改变聚合过程中AA组分的比例,可以实现对双网络聚电解质水凝胶相变行为的调控. 利用温度分辨红外光谱及二维相关分析对水凝胶的温度响应机理进行研究,结果表明体系的UCST型转变源于焓变驱动的季铵化壳聚糖与PAA链段间离子相互作用的解离和氢键作用的增强. 关于水凝胶的黏附性,涉及了丰富的分子相互作用,如PAA与基体间的氢键、季铵化壳聚糖与基体间的疏水相互作用、离子相互作用等. 二维相关红外光谱分析表明,升温相变过程中离子对解离,释放了大量解离的羧基,促使了PAA链段中羧基二聚体之间强氢键以及与季铵化壳聚糖链段羟基之间氢键的形成,提高了水凝胶的强度. 同时,水凝胶中羧基二聚体的形成有利于氨基的质子化,从而改善了组织黏附性.聚甲基丙烯酸(PMAA)在合适的水环境中也可表现出LCST型相转变[31]. 通过在PMAA水溶液中引入AlCl3等无机盐,调节盐浓度,实现了体系相转变温度的广泛可调,并构筑了具有多级结构、可实现紫外-可见-红外宽谱带光管理的新型水玻璃. 该水玻璃不仅可以可逆地切换可见光区域的透射率,阻挡紫外和红外光,还具有缺口不敏感性、自我修复断裂和划痕的功能. 借助二维相关红外光谱可对该水玻璃的动态响应机制进行解析,经分析,PMAA链段上不同化学基团在升温过程的响应次序为:α-甲基→亚甲基→羧基,表明疏水的α-甲基的脱水合是该体系相转变过程的驱动力,导致了聚合物主链的塌缩以及羧基之间氢键结构的解离. 此外,温度分辨小角X射线散射(SAXS)、微小角中子散射(VSANS)光谱证实了聚合物链塌缩引起的散射强度增加,从而产生可见光透过率的变化.一些聚电解质复合物在水溶液中也表现出热致相转变行为[32]. 通过调节典型聚电解质复合物——聚苯乙烯磺酸盐/聚二烯丙基二甲基铵在溴化钾水溶液中的浓度,同时观察到了LCST和UCST型相转变现象:低浓度下,聚电解质复合物呈现UCST型固液相转变;高浓度下,聚电解质复合物则表现为LCST型液液相分离. 基于温度分辨拉曼光谱和二维相关光谱分析,深入研究了体系中的水合效应和阴-阳离子相互作用. 研究发现,在水溶液中,聚电解质复合物的阴-阳离子相互作用呈现2种状态:直接接触型离子对(contact ion pairs, CIPs)和溶剂分离型离子对(solvent-separated ion pairs, SIPs). 聚合物浓度较低时,疏水的聚电解质链段使得阴-阳离子直接结合,CIPs占主导,而温度的升高导致了CIPs的解离,从而引起体系的UCST型转变;聚合物浓度较高时,CIPs比例低,升温导致了阴-阳离子的结合,从而引起体系的LCST型转变. 二维相关拉曼光谱分析则给出了相转变过程中的基团衍化次序,进一步揭示了聚电解质复合物两种截然不同的相转变机理:UCST型体系升温呈现出阴-阳离子相互作用逐渐减弱的解离过程,即“CIPs→SIPs→自由离子”,而LCST型体系升温呈现出阴-阳离子相互作用逐渐增强的缔合过程,即“自由离子→SIPs→CIPs”(图3). Fig. 3 2D correlation synchronous and asynchronous Raman spectra of polyelectrolyte complexes with (a) UCST- and (b) LCST-type transitions (c) Schematic illustration of the phase transition mechanisms (Reprinted with permission from Ref.[32] Copyright (2020) American Chemical Society).将温度响应聚合物引入分离膜,能够赋予膜材料温度响应功能,实现可控的物质分离[33]. 利用温敏性聚N-乙烯基己内酰胺(PVCL)和非温敏性聚乙烯基吡咯烷酮(PVP)协同稳定金属有机框架(MOF)纳米片,并进一步抽滤得到层层堆叠的温度响应纳米片复合膜. 其中PVCL提供温敏性,PVP提供支撑作用,PVCL和PVP的协同作用使得在升降温循环过程中,层间纳米孔道体积既可以同步增大和缩小,而层间距维持稳定. 所得MOF纳米片复合膜水通量及对染料截留能力具有温度敏感性. 温度升高,PVCL链塌缩使得层间纳米孔道体积增大,因而水通量增大,且升降温循环过程稳定性良好. 将尺寸相近的3种染料分子(亮绿、中性红、结晶紫)混合液进行过滤测试发现,随温度升高,尺寸较小的亮绿和中性红分子截留率下降明显高于结晶紫. 值得注意的是,对不同温度下滤液的紫外-可见光谱进行二维相关光谱分析,可以得到不同染料随温度升高的流出顺序:亮绿→中性红→结晶紫,证实了复合膜中纳米孔道尺寸随温度升高而逐渐增大. 利用二维相关红外光谱进一步对纳米片复合膜的温度响应机制进行了解析,结果显示,PVCL链段在升温过程的脱水和塌缩作为复合膜温敏行为的驱动力,降低了MOF纳米片的界面润湿性,最终导致纳米孔道的变化,而PVP链段在这一过程中并未发生明显变化,主要起到层间支撑作用(图4).Fig. 4 (a) Temperature-dependent FTIR spectra of the composite membrane (30-60 ℃). The arrows indicate the spectral variation trends at different wavenumbers (b) 2D correlation synchronous (left) and asynchronous (right) spectra of the composite membrane (c) Schematic illustration of the "smart" membrane separation performance (Reprinted with permission from Ref.[33] Copyright (2020) Springer Nature).3.2 可拉伸离子导体中复杂相互作用的揭示生命系统的生理活动与离子传导密切相关,譬如皮肤和神经纤维须通过离子传导电信号实现环境感知和运动反馈. 可拉伸离子导体是模拟弹性生物组织离子传输的重要材料,在仿生皮肤、人工肌肉、可拉伸储能、软机器人等领域取得了广泛应用.在进行可拉伸离子导体的构筑时,往往需要兼顾力学和离子传导等性能,其中涉及了丰富的分子相互作用. 本课题组围绕可拉伸离子导体,在对体系分子内/分子间相互作用机理的研究基础上,提出了一系列调控力学、电学和光学性质的分子设计. 例如:利用纳米级无定形矿物粒子和天然多糖的离子作用,调节物理交联PAA的黏弹性,所构筑的仿生皮肤可以快速自修复,且具有更高的应力响应灵敏度[34];基于AA和两性离子共聚物,选择结构匹配的离子液体,通过带电荷基团之间的离子协同效应构筑了导电纳米通道,氢键作用实现了导电通道和动态交联网络之间的协同效应,所制备的本征可拉伸导体材料透明性好、可拉伸性能突出(10000%)[35];基于聚阴离子和聚阳离子间的弱氢键相互作用构筑了一种聚离子弹性体,所得聚离子弹性体高度透明,具有接近生物组织的力学性能和感知功能,并且可以实现同步的致动和反馈效果[36];利用含氟聚离子液体与离子液体之间的离子-偶极和离子-离子相互作用,设计了一种可水下通信的光学伪装离子凝胶,该离子凝胶透明、力学性能可调、可3D打印,且具有水下自愈合、水下黏附、导离子等功能[37]. 二维相关红外光谱的优势在于从动态过程中识别体系的精细结构和复杂相互作用,因而是研究离子凝胶/弹性体中分子相互作用机制的有效手段.通过合理调控分子间/内相互作用,设计制备了一种基于天然小分子α-硫辛酸(α-thioctic acid, TA)的可涂覆离子凝胶油墨(图5)[38]. 在离子液体1-乙基-3-甲基咪唑硫酸乙酯([EMI][ES])存在的条件下,TA室温即可进行浓度诱导的自发开环聚合,得到稳定、透明、高拉伸且自愈合的离子凝胶弹性体. 该弹性体易溶于乙醇,因而能够方便地涂覆到任意表面,赋予涂覆体稳定的离子导电能力和应变感知功能. 利用红外光谱等手段探讨了离子凝胶中离子液体对聚硫辛酸(polyTA)的稳定机制:相比于纯的polyTA体系,离子凝胶的COOH伸缩振动区域在1734 cm-1出现了明显的肩峰,而离子液体的S=O伸缩振动峰在离子凝胶中呈现了明显的红移,表明polyTA的羧基与硫酸乙酯阴离子形成了COOH… [ES]氢键. 分子动力学模拟结果表明了COOH… [ES]氢键的热力学稳定性,同时该氢键能够有效降低polyTA的势能. 因此,离子液体主要通过阴离子ES与polyTA基间形成强氢键而稳定polyTA. 二维相关红外光谱则揭示了离子凝胶升温过程不同化学基团的响应次序:COOH… [ES]氢键→羧酸二聚体→自由羧基,说明COOH… [ES]氢键对温度变化最敏感,进一步证实了COOH… [ES]氢键对于稳定polyTA离子凝胶的重要作用. Fig. 5 (a) Schematic illustration of the COOH[ES] H-bonding in the ionogel (b) ATR-FTIR spectral comparison among ionogel, [EMI][ES] and neat polyTA (c) Temperature-variable FTIR spectra of the ionogel in the C=O stretching region from 25 °C to 151 °C Perturbation-correlation moving window (d) and 2D correlation synchronous and asynchronous spectra (e) generated from (c). (Reprinted with permission from Ref.[38] Copyright (2021) Wiley).受指纹结构启发,构筑了一种具有共形和可重复编辑褶皱结构的本征可拉伸离子导电芯鞘纤维[39],其中,纤维芯层为离子凝胶弹性体,鞘层为氟橡胶,芯鞘界面借助共价交联网络和离子-偶极相互作用实现协同拓扑互锁和物理黏附. 经过表面褶皱结构的优化,该离子纤维拉伸应变感知灵敏度(gauge factor)可提升至10以上,超过了绝大多数可拉伸离子导体应变传感器. 利用红外光谱对离子凝胶芯层的分子相互作用进行研究,发现其中涉及了离子液体阳离子咪唑环上C―H与聚合物侧基乙氧基间的氢键、聚合物链段C=O间的偶极-偶极相互作用、离子液体阴-阳离子间的弱静电相互作用等,而这些都对离子凝胶的高拉伸行为做出了重要贡献. 基于对芯层和鞘层力学性能的研究,发现表面褶皱形成的主要原因在于,高模量的氟橡胶鞘层弹性回复率显著低于离子凝胶芯层,在应变回复过程中造成了芯层和鞘层的界面失稳. 随着预应变的增加,弹性回复率差异变大,从而导致更加密集的褶皱结构. 此外,形成的表面褶皱可通过加热至60 ℃完全消除,从而赋予纤维可重复编辑褶皱的能力. 二维相关红外光谱揭示了离子凝胶芯层高温下残余应变的消除主要源于聚合物链段C=O间偶极-偶极相互作用的减弱和构象重排,而氟橡胶鞘层由C―F间偶极-偶极相互作用锚定的链构象也可以通过加热消除.通过在强氢键交联的PAA网络中引入熵驱动的弱交联两性离子超分子网络,产生竞争机制,设计制备了一系列透明、抗冻、保湿、黏附、高拉伸、高回弹、自愈合、应变硬化、导质子、可重复加工等综合性能优异的离子皮肤(图6)[40]. 不同于传统水凝胶和离子凝胶,该离子弹性体不含大量溶剂,仅含有少量达到吸湿平衡的水分子,这使得分子间的羧酸二聚体氢键足以交联PAA分子链而形成强交联网络,而弱交联的两性离子超分子网络则提供柔性. 通过红外光谱、核磁共振谱和力学松弛等实验探讨了这一二元网络体系中的分子相互作用. 其中,具有较低pKa值的两性离子的存在使得PAA轻度去质子化,游离的质子是主要载流子. 去质子化的PAA与两性离子的阳离子端也可以发生离子缔合. 利用变温红外光谱并结合二维相关光谱分析,验证了体系中的3种主要分子相互作用,并根据它们对于温度的响应顺序判别了其结合强度,即PAA链段羧酸二聚体氢键 PAA-甜菜碱离子相互作用 甜菜碱-甜菜碱离子相互作用,这一光谱表征结果为该离子皮肤强弱协同竞争网络的分子设计提供了重要依据. Fig. 6 (a) Temperature-variable FTIR spectra of PAA/betaine ionic elastomer upon heating (b) 2D correlation synchronous and asynchronous spectra generated from (a) FTIR (c) and 1H-NMR (d) spectra of PAA, betaine, and PAA/betaine (e) Schematic illustration of PAA/betaine elastomer and the order of interaction strength among the three main interacting pairs (Reprinted with permission from Ref.[40] Copyright (2021) Springer Nature).3.3 小分子在聚合物基质中的扩散聚合物生产和加工的许多工序都涉及小分子物质在聚合物基体的扩散,研究这类扩散行为具有重要的理论和实践意义. ATR-FTIR光谱可对小分子在聚合物基质中的扩散过程进行实时、原位、快速、多组分检测,能够同时获取扩散系数和分子层面相互作用等信息. 扩散装置示意图如图7所示,聚合物基体处于ATR晶体和扩散物质之间,当扩散物质从聚合物基体的上表面扩散至下表面时即可被检测到. 随着时间的增加,与扩散物质相关的特征吸收峰强逐渐增大直至扩散平衡(扩散谱图,图7(b)). 以扩散时间为横坐标、扩散物质特征吸收峰强度/面积为纵坐标作图,即可得到扩散曲线(图7(c)). 结合二维相关光谱分析,可以提供动态扩散过程结构与相互作用的变化信息,有助于解析扩散机制[41~45].Fig. 7 (a) Schematic illustration of the diffusion experiments by ATR-FTIR spectroscopy (b) typical diffusion spectra (c) a typical diffusion curve.基于朗伯比尔定律和菲克扩散模型,Fieldson等[46]建立了基于ATR-FTIR光谱测试计算扩散系数的公式:其中这里,At为扩散时间t时,特征红外吸收峰的强度或面积;A∞为扩散达到平衡时,特征红外吸收峰的强度或面积;L为聚合物薄膜基体的厚度;D为扩散剂的扩散系数;γ为光波在聚合物基体中渗透深度的倒数,可表示为:其中,θ (θ = 45o)为红外光的入射角;n1和n2分别为聚合物和ATR晶体的折光指数;λ为红外光的波长. 基于以上扩散方程对ATR-FTIR光谱测试得到的扩散曲线进行拟合,即可得到相关扩散系数. 此外,根据曲线拟合情况可以判断该扩散过程的扩散模型.利用时间分辨ATR-FTIR光谱并结合二维相关光谱分析技术对水分子在乙基纤维素(EC)基薄膜中的扩散行为进行系统研究[47]. 分析表明,水分子在EC中的扩散行为符合菲克扩散模型,通过对扩散曲线的拟合计算得到了相关的扩散系数. 此外,探讨了EC中增塑剂(柠檬酸三乙酯)含量对水分子扩散行为的影响,结果表明,增塑剂的添加不影响水分子的扩散模型,主要起到加速水分子扩散的作用,这主要源于增塑剂的加入改善了EC链的活动性而提高了EC基体的自由体积(free volume). 利用二维相关光谱对水分子羟基伸缩振动区域扩散谱图进行解析,观察到在整个扩散过程中,主要存在着4种类型的水分子,即本体水(强氢键作用)、团簇水(中等强度氢键作用)、相对自由的水分子(弱氢键作用)以及自由的水分子(极弱氢键作用). 依据Noda规则,判别出不同状态水分子扩散的先后顺序:团簇水→本体水→相对自由的水分子或自由的水分子,表明扩散首先来自体积较小、相对弱氢键结合的团簇水,其次才是大量的本体水,而随着扩散过程的进行,部分水分子与聚合物基体相互作用而脱离团簇水或本体水,产生了(相对)自由的水分子.EC被广泛用作药物包衣材料以实现药物缓释的功能,利用ATR-FTIR光谱对药物分子在EC基薄膜中的扩散行为进行实时监测可以有效模拟这一药物缓释过程(图8),从而为EC基药物包衣材料的配方优化提供理论指导[48]. 扩散谱图直观呈现了体系中各组分的变化情况,包括水分子(1637 cm-1)和药物分子(1569 cm-1)特征吸收峰强度的上升,增塑剂(1737 cm-1)特征吸收峰强度的下降等,表明水分子和药物分子在EC基薄膜中的扩散以及薄膜中增塑剂的部分溶解. 定量分析结果表明,扩散主要包含3个阶段:(A)水分子扩散;(B) EC膜吸水饱和,水扩散停止并溶解EC基体中的致孔剂;(C) 随着致孔剂的溶解,EC薄膜中形成孔道,使得药物分子和水分子共同扩散,同时增塑剂溶解. 二维相关红外光谱分析结果进一步证实了C阶段的各组分变化顺序:水分子扩散→药物分子扩散→增塑剂溶解,并且显示药物分子始终处于水合状态. 此外,通过改变药物分子的水溶性、致孔剂的种类以探讨膜配方对扩散行为的影响,结果表明随着致孔剂水溶性的增加和/或药物分子水溶性的降低,B阶段将缩短甚至消除. Fig. 8 (a) Time-resolved ATR-FTIR spectra collected during the water and drug diffusion (b) 2D correlation synchronous and asynchronous spectra during the diffusion of Stage C (c) Schematic illustration of water and drug diffusion across the EC-based film (Reprinted with permission from Ref.[48] Copyright (2015) Elsevier).氢氧化物/尿素是溶解纤维素的重要组合,其中尿素可稳定纤维素的疏水部分,有利于形成包合物从而促进纤维素的溶解. 在分子层面上,尿素溶液对纤维素的作用机理尚不明确. 采用ATR-FTIR光谱并结合二维相关光谱衍生的外扰相关移动窗口(perturbation-correlation moving window,PCMW)技术研究了不同浓度尿素水溶液(0,20 wt%、40 wt%和50 wt%)在黏胶纤维膜中的扩散行为,在分子水平揭示了尿素溶液的动态扩散行为以及与黏胶纤维的相互作用机制[49]. 从扩散谱图的变化规律以及对应的扩散曲线看,尿素溶液的扩散过程可大致分为2个步骤,水分子首先通过黏胶纤维膜,随后带动尿素分子一起通过. PCMW谱图显示,尿素浓度越高,尿素分子扩散滞后现象越明显. 根据菲克扩散模型,尿素分子在黏胶纤维膜的扩散系数随尿素浓度的增加而减小. 在红外光谱中,特征谱峰出现位移表明相应官能团相互作用的变化. 基于扩散过程Amide Ⅲ(尿素)和CH2-O(6)H伸缩振动(纤维素)的峰位移变化趋势,尿素水溶液在黏胶纤维中的扩散过程可以概括为:首先水分子破坏黏胶纤维膜无定形区的氢键网络,与羟基形成新的纤维素-水氢键,随后尿素分子在水分子的“桥连”作用下形成纤维素-水-尿素氢键,从而间接作用于纤维素. 低浓度下,水分子相对含量较大,可以快速打开扩散通道带动尿素分子通过黏胶纤维膜. 而高浓度下,尿素分子发生聚集且固定了大量水分子,从而在宏观上延缓了尿素溶液的扩散.热转移印花是纺织品印花方法之一,本质上是分散染料向聚酯纤维动态扩散的过程. 借助ATR-FTIR光谱对分散红9 (DR 9)在聚对苯二甲酸乙二醇酯(PET)薄膜中的扩散过程进行了原位跟踪,模拟了热转移印花过程,并结合二维相关光谱探讨了分散染料-分散染料、分散染料-PET相互作用机制,在分子水平上阐释了其扩散机理(图9)[50]. DR 9在PET薄膜中的扩散过程符合菲克扩散模型. 温度越高,扩散速度越快,这主要归因于:(1) 温度升高导致了PET基体自由体积的增加和分子链热运动的增强;(2) DR 9在高温下分子运动的增强. 此外,将不同温度下的扩散系数按照Arrhenius公式进行线性拟合,可以计算得到DR 9在PET中扩散活化能为15.33 kJ/mol. 通过对扩散过程中不同阶段的红外谱图进行对比,观察到了体系中存在丰富的分子间/内相互作用,包括PET和DR 9的C=O基团间偶极-偶极相互作用、芳香基团间π-π相互作用以及DR 9分子内氢键等. 二维相关红外光谱分析进一步细化了扩散体系中不同化学基团的分子间/内相互作用及其在扩散过程中的变化情况. 高温下,随着DR 9分子热运动增强,DR 9分子之间的相互作用减弱. 借助DR 9和PET中C=O基团之间的偶极-偶极相互作用,DR 9扩散进入PET基体. 在扩散过程中,DR 9中形成了较强的分子内氢键,从而提高了DR 9的平面性,促进了扩散过程. 随着越来越多的DR 9分子扩散到PET基体中,DR 9和PET的芳香基团之间的π-π相互作用成为主导,DR 9的分子内氢键减弱. Fig. 9 (a) Time-resolved ATR-FTIR spectra and (b) 2D correlation synchronous and asynchronous spectra of DR 9 diffusion in PET at 140 ℃ (c) Schematic diagram of DR 9 diffusion into PET (Reprinted with permission from Ref.[50] Copyright (2020) American Chemical Society).采用时间分辨ATR-FTIR光谱对不同温度下碳酸丙烯酯(PC)-双三氟甲磺酰亚胺锂(LiTFSI)在聚偏氟乙烯-六氟丙烯共聚物(P(VDF-HFP))中的扩散行为进行了原位监测,同时获得了凝胶聚合物电解质中各扩散组分的扩散系数和分子层面相互作用信息[51]. 基于PC中C=O伸缩振动区域的二阶导数分析,推断出PC在凝胶电解质主要存在四种状态,即与P(VDF-HFP)发生偶极-偶极相互作、PC分子间发生偶极-偶极相互作用、与锂离子发生强离子-偶极相互作用、与锂离子发生弱离子-偶极相互作用. 同时,LiTFSI参与的分子相互作用也得以识别,包括锂离子与PC中C=O之间的离子-偶极相互作用,锂离子与P(VDF-HFP)中C―F之间的离子-偶极相互作用、TFSI-的溶剂化作用等. 扩散过程中,首先是PC分子以溶剂团簇的形式扩散进入P(VDF-HFP),PC分子中的C=O与P(VDF-HFP)中的C―F发生偶极-偶极相互作用,一定程度减弱了P(VDF-HFP)聚合物链间的偶极-偶极相互作用,从而有利于锂盐的扩散. 随后,借助锂离子与C=O的离子-偶极相互作用,锂离子随着PC分子扩散进入P(VDF-HFP),TFSI-在扩散过程中也一直处于溶剂化状态. 这里,PC分子既充当了增塑剂的角色,同时也是离子(包括阴离子和阳离子)扩散的载体. 本工作在分子水平上揭示了PC-LiTFSI在P(VDF-HFP)的传导机制,对高性能凝胶聚合物电解质的结构设计和性能优化具有一定的指导意义.3.4 天然高分子的结构表征海藻酸钠(SA)作为一类天然多糖,生产成本低、无毒且具有良好的生物相容性、可降解性,在食品工业、制药、纺织印染等领域得到了广泛应用. 随着实验室和工业对SA的日趋重视,理解SA内部的氢键结构也变得越发重要. 利用红外光谱对SA升温过程特征基团的变化进行原位监测,结合二维相关光谱等分析手段从分子水平研究了SA体系的相互作用机制,探讨了温度扰动下SA分子间/内、SA与水分子间氢键结构的演变历程[52]. 研究发现,加热过程可分为30~60 ℃和60~170 ℃ 2个阶段:第一阶段为弱氢键结合的水分子脱除,第二阶段为强氢键结合的水分子脱除. 二维相关红外光谱结果表明:30~60 ℃区间内,随脱水过程发生,SA与水分子的氢键逐步断裂,SA中C―OH和COO-基团逐渐参与形成分子间/内氢键(O3H3⋯O5和O2H2⋯O=C―O-),因此水分子的存在一定程度破坏了SA中原有的氢键结构;60~170 ℃区间内,强结合水脱除,SA与水分子的氢键进一步断裂,同时SA分子间/内氢键相互作用逐步减弱,出现了部分相对自由的C―OH和COO-基团(图10). 由于相对自由的COO-比C―OH更早出现,可以推测C―OH形成的分子间/内氢键相互作用比COO-更强.Fig. 10 2D synchronous and asynchronous spectra of the SA film during heating between (a) 30-60 °C and (b) 60-170 °C (c) Schematic illustration of the heat-induced hydrogen bonding transformation in the SA film[52] (Reprinted with permission from Ref.[52] Copyright (2019) Elsevier).多元羧酸与纤维素的羟基反应,能使纤维素大分子间形成立体的交联网络结构,从而赋予棉纤维织物抗皱性能. 1,2,3,4-丁烷四羧酸(BTCA)作为一类典型的用于棉纤维织物抗皱整理的多元羧酸,其与纤维素的酯化过程受到了广泛关注,但其中关于分子水平相互作用机制及动态反应机理仍不清晰. 利用FTIR光谱对加热过程中纤维素与BTCA在催化剂次亚磷酸钠(SHP)作用下的酯化反应过程进行原位跟踪,并借助二维相关光谱分析技术探讨了该反应的分子机理,重点关注了分子层面相互作用机制以及反应全过程中的化学基团转变历程[53]. 分析表明,室温下,体系中的O―H和C=O等极性基团有强氢键相互作用. SHP存在时,碱金属离子(Na+)与羧基反应并将其转化为相应的羧酸盐,从而一定程度削弱了BTCA间的氢键相互作用. 在30~100 ℃的加热过程中,体系中的氢键部分断裂,导致一些O―H和C=O处于相对自由的状态. 这里,SHP的存在和加热过程都会导致体系中氢键相互作用的减弱,从而使相应的化学基团更自由,有利于酸酐生成和酯化反应. 当加热至100 ℃以上后,羧酸盐和自由羧酸开始脱水形成环酐. 一旦形成环酐,就会与纤维素大分子链上的O―H反应生成酯. 通过逐步成酐和酯化反应过程,BTCA实现了对纤维素的交联. 该结果对多元羧酸的抗皱整理工艺优化及寻找更有效的多元羧酸类抗皱整理剂和催化剂具有一定的指导作用.4 总结与展望本文主要介绍了二维相关光谱的基本原理、实验和分析技巧等,并结合具体的体系(如温度响应高分子、可拉伸离子导体、小分子在聚合物中的扩散过程、天然高分子等),简述了二维相关光谱在高分子表征中的应用. 这里,二维相关光谱不仅能够有效鉴别高分子体系涉及的丰富相互作用,还能提供外扰作用下动态过程发生的分子机制. 相关研究结果一方面有助于启发新型功能高分子材料的结构设计,另一方面也可以为实际工艺过程的配方优化和参数调整提供指导.二维相关光谱作为一种先进的光谱分析手段,在高分子材料体系的表征中得到了越来越多的关注. 随着高分子材料涉及的体系越来越复杂、功能越来越强大,这为二维相关光谱的应用提供了更多的机遇,但同时也带来了更多的挑战. 在后续的研究工作中,二维相关光谱分析可以重点关注以下几方面:(1) 光谱手段的多样性. 目前关于二维相关光谱在高分子体系中的应用主要是基于中红外光谱,关注的是分子层面相互作用信息. 一方面,中红外光谱也有一定的局限性,例如低浓度溶液体系信号弱、水的吸收峰干扰严重等. 对于中红外光谱难以表征的体系,可以尝试其他分子光谱手段,如拉曼光谱、近红外光谱等,开展二维相关光谱分析. 另一方面,其他光谱手段,包括荧光光谱、圆二色谱、紫外-可见吸收光谱、X射线衍射谱等,都可以进行二维相关光谱分析,以获取多层面丰富的结构信息. 目前,这些光谱在处理二维相关分析时,大部分因信噪比低而导致噪音被显著放大,使得结构解析变得困难,如何有效解决这一问题是丰富二维相关分析光谱手段的关键.(2) 外扰变量的丰富性. 时间、温度便于控制,是目前获取动态光谱最常用的外扰变量. 然而,影响高分子结构和性能的因素是多种多样的,例如湿度变化能够引起高分子力学性质的改变、紫外光照射可以引起高分子的老化等,尤其是刺激响应高分子,可以对温度、压力、电场、磁场、pH、浓度等丰富的外扰产生响应,引起物理或化学性质的变化. 最近,Li等[54]利用二维相关红外光谱研究了乙醇诱导聚丙烯酰胺/Pluronic 127水凝胶相分离的机理,获取了氢键解离和无定形-结晶转变等信息. 因此,利用二维相关光谱探讨不同刺激下高分子结构的演变机制,将进一步拓宽二维相关光谱的应用范围. 需要注意的是,对于测试过程无法原位施加的外扰变量,应尽量避免其他因素改变而引起的光谱变化,否则将影响二维相关光谱分析结果的真实性和可靠性.(3) 多种分析手段的关联. 一方面,通过二维相关光谱交叉谱的计算和解析,可以将不同分析手段所得结果进行关联,这能够帮助理解高分子不同层面结构的内在联系. 另一方,二维相关光谱分析结果涉及丰富的相互作用和结构变化,经过与其他分析表征手段的结果进行比对和相互验证,可有效加深人们对二维相关光谱分析结果的理解. 参考文献1Ernst R R, Bodenhausen G, Wokaun A. Principles of Nuclear Magnetic Resonance in one and Two Dimensions. Oxford: Clarendon Press, 19872Noda I, Dowrey A, Marcott C, Story G, Ozaki Y. Appl Spectrosc, 2000, 54(7): 236A-248A. doi:10.1366/0003702001950454 3Noda I. J Am Chem Soc, 1989, 111(21): 8116-8118. doi:10.1021/ja00203a008 4Noda I. Appl Spectrosc, 1990, 44(4): 550-561. doi:10.1366/0003702904087398 5Noda I. Appl Spectrosc, 1993, 47(9): 1329-1336. doi:10.1366/0003702934067694 6Noda I. Anal Sci, 2007, 23(2): 139-146. doi:10.2116/analsci.23.139 7Park Y, Jin S, Noda I, Jung Y M. J Mol Struct, 2020, 1217: 128405. doi:10.1016/j.molstruc.2020.128405 8Sun S, Tang H, Wu P, Wan X. Phys Chem Chem Phys, 2009, 11(42): 9861-9870. doi:10.1039/b909914j 9Kim Y J, Matsunaga Y T. J Mater Chem B, 2017, 5(23): 4307-4321. doi:10.1039/c7tb00157f 10Chilkoti A, Dreher M R, Meyer D E, Raucher D. Adv Drug Deliv Rev, 2002, 54(5): 613-630. doi:10.1016/s0169-409x(02)00041-8 11Weber C, Hoogenboom R, Schubert U S. Prog Polym Sci, 2012, 37(5): 686-714. doi:10.1016/j.progpolymsci.2011.10.002 12Tang L, Wang L, Yang X, Feng Y, Li Y, Feng W. Prog Mater Sci, 2021, 115: 100702. doi:10.1016/j.pmatsci.2020.100702 13Sun B, Lin Y, Wu P, Siesler H W. Macromolecules, 2008, 41(4): 1512-1520. doi:10.1021/ma702062h 14Sun S, Wu P. Macromolecules, 2010, 43(22): 9501-9510. doi:10.1021/ma1016693 15Sun S, Wu P. J Phys Chem B, 2011, 115(40): 11609-11618. doi:10.1021/jp2071056 16Wang H, Sun S, Wu P. J Phys Chem B, 2011, 115(28): 8832-8844. doi:10.1021/jp2008682 17Sun B, Lai H, Wu P. J Phys Chem B, 2011, 115(6): 1335-1346. doi:10.1021/jp1066007 18Sun S, Wu P. Macromolecules, 2013, 46(1): 236-246. doi:10.1021/ma3022376 19Zhang B, Tang H, Wu P. Macromolecules, 2014, 47(14): 4728-4737. doi:10.1021/ma500774g 20Hou L, Wu P. Soft Matter, 2014, 10(20): 3578-3586. doi:10.1039/c4sm00282b 21Hou L, Wu P. Soft Matter, 2015, 11(14): 2771-2781. doi:10.1039/c5sm00026b 22Sun W, An Z, Wu P. Macromolecules, 2017, 50(5): 2175-2182. doi:10.1021/acs.macromol.7b00020 23Hou L, Ma K, An Z, Wu P. Macromolecules, 2014, 47(3): 1144-1154. doi:10.1021/ma4021906 24Li T, Tang H, Wu P. Soft Matter, 2015, 11(10): 1911-1918. doi:10.1039/c4sm02812k 25Sun S, Hu J, Tang H, Wu P. J Phys Chem B, 2010, 114(30): 9761-9770. doi:10.1021/jp103818c 26Sun S, Wu P. Chinese J Polym Sci, 2017, 35(6): 700-712. doi:10.1007/s10118-017-1938-1 27Sun Shengtong(孙胜童), Wu Peiyi(武培怡). Materials Science and Technology(材料科学与工艺), 2017, 25(1): 1-9. doi:10.11951/j.issn.1005-0299.20160386 28Lei Z, Wu P. Nat Commun, 2018, 9(1): 1134. doi:10.1038/s41467-018-03456-w 29Lei Z, Wu P. ACS Nano, 2018, 12(12): 12860-12868. doi:10.1021/acsnano.8b08062 30Shi X, Wu P. Small, 2021, 17(26): 2101220. doi:10.1002/smll.202101220 31Lei Z, Wu B, Wu P. Research, 2021, 2021: 4515164. doi:10.34133/2021/4515164 32Ye Z, Sun S, Wu P. ACS Macro Lett, 2020, 9(7): 974-979. doi:10.1021/acsmacrolett.0c00303 33Jia W, Wu B, Sun S, Wu P. Nano Res, 2020, 13(11): 2973-2978. doi:10.1007/s12274-020-2959-6 34Lei Z, Wang Q, Sun S, Zhu W, Wu P. Adv Mater, 2017, 29(22): 1700321. doi:10.1002/adma.201700321 35Lei Z, Wu P. Nat Commun, 2019, 10(1): 3429. doi:10.1038/s41467-019-11364-w 36Lei Z, Wu P. Mater Horiz, 2019, 6(3): 538-545. doi:10.1039/c8mh01157e 37Yu Z, Wu P. Adv Mater, 2021, 33(24): 2008479. doi:10.1002/adma.202008479 38Wang Y, Sun S, Wu P. Adv Funct Mater, 2021, 31(24): 2101494. doi:10.1002/adfm.202101494 39He C, Sun S, Wu P. Mater Horiz, 2021, 8(7): 2088-2096. doi:10.1039/d1mh00736j 40Zhang W, Wu B, Sun S, Wu P. Nat Commun, 2021, 12(1): 4082. doi:10.1038/s41467-021-24382-4 41Shen Yi(沈怡), Peng Yun(彭云), Wu Peiyi(武培怡), Yang Yuliang(杨玉良). Progress in Chemstry(化学进展), 2005, (3): 499-513. doi:10.3321/j.issn:1005-281X.2005.03.016 42Liu M, Wu P, Ding Y, Chen G, Li S. Macromolecules, 2002, 35(14): 5500-5507. doi:10.1021/ma011819f 43Tang B, Wu P, Siesler H W. J Phys Chem B, 2008, 112(10): 2880-2887. doi:10.1021/jp075729+ 44Wang M, Wu P, Sengupta S S, Chadhary B I, Cogen J M, Li B. Ind Eng Chem Res, 2011, 50(10): 6447-6454. doi:10.1021/ie102221a 45Lai H, Wang Z, Wu P, Chaudhary B I, Sengupta S S, Cogen J M, Li B. Ind Eng Chem Res, 2012, 51(27): 9365-9375. doi:10.1021/ie300007m 46Fieldson G T, Barbari T A. Polymer, 1993, 34(6): 1146-1153. doi:10.1016/0032-3861(93)90765-3 47Hou L, Feng K, Wu P, Gao H. Cellulose, 2014, 21(6): 4009-4017. doi:10.1007/s10570-014-0458-1 48Feng K, Hou L, Schoener C A, Wu P, Gao H. Eur J Pharm Biopharm, 2015, 93: 46-51. doi:10.1016/j.ejpb.2015.03.011 49Dong Y, Hou L, Wu P. Cellulose, 2020, 27(5): 2403-2415. doi:10.1007/s10570-020-02997-y 50Yan L, Hou L, Sun S, Wu P. Ind Eng Chem Res, 2020, 59(16): 7398-7404. doi:10.1021/acs.iecr.9b07110 51Li H, Hou L, Wu P. Chinese J Polym Sci, 2021, 39(8): 975-983. doi:10.1007/s10118-021-2571-6 52Hou L, Wu P. Carbohydr Polym, 2019, 205: 420-426. doi:10.1016/j.carbpol.2018.10.091 53Hou L, Wu P. Cellulose, 2019, 26(4): 2759-2769. doi:10.1007/s10570-019-02255-w 54Li Y, Wang D, Wen J, Liu J, Zhang D, Li J, Chu H. Adv Funct Mater, 2021, 31(22): 2011259. doi:10.1002/adfm.202011259 《高分子学报》高分子表征技术专题链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304原文链接:http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2021.21362DOI:10.11777/j.issn1000-3304.2021.21362























 我要推广仪器
我要推广仪器
 下载APP
下载APP