有这么一种流动相,甲醇:水:三乙胺:乙酸=600:400:1:1那么加三乙胺和乙酸的作用是什么?
通常液相色谱分析中,在流动相中加乙酸和三乙胺,大家来谈谈,在什么情况下需要加乙酸,什么情况下加三乙胺,加多少是怎么控制的?
二乙烯三胺五乙酸浸提土壤的实验为什么我做其他的都没问题,只有铁和锰的空白特别高
如何检测哌嗪、乙二胺、三乙烯二胺、二乙烯三胺、氨乙基哌嗪、二甲苯、均三甲苯?
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先 补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论跟上次的问题一样,上次结贴太匆忙了,再次征集。感谢上次renture/jiangyunjun3/melu的帮助,找到一个分光法测痕量氮川的方法,测量范围是0.01~0.12ug/mL,但我们的样品是含量大概有0~10%左右的氮川,是否能够通过稀释来使用上面的分光方法还不知道。现在想征集一个能够分析常量氮川的方法,希望大家踊跃发言。
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先[em0813] [em0813] [em0813]补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论
怎么分离亚氨基二乙酸和氨基三乙酸,样品中氨基三乙酸含量少
如何检测无水哌嗪和三乙烯二胺的水分?
在用液相色谱分析某些酸性药品时,有的时候会在流动相中加入三氟乙酸,加入三氟乙酸的目的是什么啊?有时候还会在流动相中加入二乙胺,目的是什么啊?仅仅是要调节pH值么?还有就是,二乙胺可以用三乙胺代替么?哪位高手给解答一下啊,具体点的。
GC,我用色谱柱TG-624 检测三乙胺、乙酸异丙酯和庚烷的混合样,分离总是不好。该如何调节?
行标YC 144-2008中规定三乙酸甘油酯纯度测定时,气相进样口温度为250℃。但我查到的三乙酸甘油酯沸点为:258-260 °C(lit.)℃。进样口的温度设置原则不应该是要高于被分析物的沸点,确保所有分析物经过进样口进样后能够完全气化吗?想寻求大家的解答
原子吸收测定三乙酸甘油酯、沒食子酸丙酯中铅、镉的含量?那位做过,我现在查的沒食子酸丙酯中铅的测定按照GB/T5009.75测定,请问哪里有标准下载。三乙酸甘油酯、沒食子酸丙酯中铅镉的含量都是微量
师傅您好请问用wadnacap1和wandacap5两种型号的色谱柱那种色谱柱可以测定乙二胺,哌嗪,三乙烯二胺,二乙烯三胺,AEP,岛津2014C各参数值是多少?
[color=#444444]各位大神给点建议,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]所用的流动相是三氟乙酸和三乙胺配制的缓冲液,来检测发酵液中的有机酸,能不能给点建议,具体怎样配制此缓冲液?[/color]
今天用液相色谱仪检测依地酸钙钠中的氨基三乙酸,用的是安捷伦的C8柱子,流动相为0.01mol/L的氢氧化四丁基铵:甲醇=90:10,溶剂为硝酸铜,可是压力很不稳定,好不容易平衡后,一进样,压力瞬间飘很高,在走样过程中逐渐下降,走一针过后,基线很不稳定,感觉柱子里面冲出来很多东西,有没有做过这个品种的前辈们啊,知道我一下吧,怎么改进?
我求解,三乙胺残留量气相检测需要注意什么?三乙胺好检测吗?检测它难点在哪儿?色谱柱选择针对该应用有什么讲究吗?极性柱和非极性柱哪种好些?试过HP-innowax柱,水做溶剂,顶空进样(平衡105度,瓶加压30psi),峰形不好,拖尾。峰面积RSD有时超过15%。跟它一同进柱子的丙酮、乙酸乙酯等组分,都表现良好。FID检测器对它的响应好不好,够不够灵敏?顶空瓶中水溶液中浓度为9ppm,agilent 7697顶空,分流比5:1,峰面积15左右,正常不?有人说三乙胺溶液中应加少量氢氧化钠处理,再做效果好,可行不?
[font=宋体][size=24px][color=#3366ff][b]仪器是ICP 赛默飞6300 .[/b][/color][/size][/font][size=24px][color=#3366ff][b][font=宋体]方法需要用到浸提液:[/font][font=宋体]三乙醇胺和[/font][font=宋体]二乙烯三胺五乙酸,里面有有机物![/font][/b][/color][/size][font=宋体][size=24px][b][color=#3366ff]按照标准的意思,它是直接进样的。[/color][color=#ff0000]用ICP是否可以直接测????[/color][/b][/size][/font][font=宋体][size=24px][color=#3366ff][b]哪位大神做过这个方法?需要改变功率吗?需要配制有机物进样装置吗?[/b][/color][/size][/font][font=宋体][size=24px][color=#3366ff][b]还是可以直接进样不熄火???如果不能直接进样,应该怎么做???[/b][/color][/size][/font][font=宋体][size=24px][color=#3366ff][b]O(∩_∩)O谢谢大家的回复[/b][/color][/size][/font]
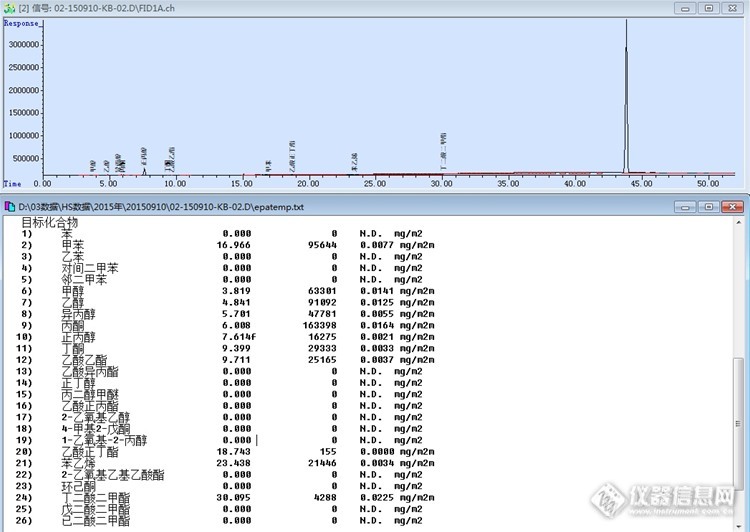
安谱三乙酸甘油酯的使用心得 我们实验室一直在测烟标中的VOC,参照烟标YC/T 207-2014的方法来检测,YC/T207-2014同YC/T207-2006相比培加了10种管控物质,同时对溶剂杂质也有要求,基于方法的加严,为了达到更好的检测数据,对检测用的试剂自然而然的加严了,所以,就采购的安谱药典级的三乙酸甘油酯。以前我们使用的某品牌的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819191794_01_2769262_3.jpg现在我们使用的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819194869_01_2769262_3.jpg现就两种不同的三乙酸甘油酯,做个对比测试,选择用新的20ml顶空进样瓶,然后分别移取不同品牌的三乙酸甘油酯到顶空进样瓶中,封盖,然后用顶空加气相(带FID检测器)检测,检测结果如下:某品牌的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223603_01_2769262_3.jpg安谱药典级的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223175_01_2769262_3.jpg两种品牌试剂的检测结果汇总表:http://ng1.17img.cn/bbsfiles/images/2015/09/201509181923_566740_2769262_3.jpg综上,安谱药典级的三乙酸甘油酯优于某品牌的三乙酸甘油酯,安谱药典级的三乙酸甘油酯值得版友们拥有。
请问EDTA中氨基三乙酸的含量,不合格的批次多吗?
三乙胺和磷酸反应吗,如果反应那生成物遇到三氟乙酸会出现什么情况?谢谢!
要测一个原料药中三乙胺的残留。仪器是安捷伦的6890N,7694E,条件如下:色谱柱HP-1,柱温100,进样口250,分流比1:1,柱压7.7psiFID250顶空平衡30min,温度70/100/110三乙胺浓度16ug/ml(用水溶),取2ml至10ml顶空瓶我的问题是我进了三乙胺后跟了一针空白,三乙胺的位置有出峰,再跟一针空白就没有了,我试着做进样精密度,连续进了三针峰面积从29涨到了40,RSD非常差。而且换了DB-624的柱子,这个问题还是存在,哪位大侠指点一下!
我手工过柱子,分离约15克的含N化合物,点板发现拖尾严重,流动相加入三乙胺或者氨水点板,就收到明显效果,不再拖尾。所以过柱子的时候,石油醚-乙酸乙酯流动相中需要加入氨水或者三乙胺。但是,加入氨水的话,氨水含有很多水,跟石油醚等互不相溶;加入三乙胺,师兄说很难蒸发除掉,会混在我的产物里面。因此,求助于各位高手,我过柱子的时候,到底该加什么?
在对黄连进行薄层色谱的时候,用环己烷:乙酸乙酯:异丙醇:甲醇:水:三乙胺(3:3.5;1:1.5:0.5:1)进行展开,但是没有展开完,就感觉有二次展开的情况,想问问出现这种情况的原因,和解决方法
原子荧光测定三乙酸甘油酯、沒食子酸丙酯中砷、汞的含量?含量是微量的,样品是采用湿法消解还是采用微波消解?
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
[color=#444444]三乙胺-丙酮-盐的混合液,三乙胺的含量在5%以下,请问有什么好的方法可以测定三乙胺的含量,在实验室用氢火焰毛细柱打可以出峰,但是如果含量特别低时担心[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]打不出,导致微量三乙胺检测不出来。查文献有溴酚蓝分光光度法、红外色谱,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]、电位返滴定法,以及混合液放入盐酸,用过量NaOH滴定未反应酸,溴酚蓝做指示剂等方法。请问这几种方法哪个比较准确,并且好操作,谢谢各位了![/color]
我是A的一名用户,上次有个研究生做项目连续用半个月50mM三乙胺水溶液做项目(先前休假不知道),后来我接仪器后调谐过不过后报修。A的工程师来后发现是三乙胺搞鬼,让我用0.5~0.8%乙酸/甲酸水冲洗仪器手动改了下参数就走人。后来我就配了4L.8%乙酸1mL/min冲洗,开始打到MS本底有6M,连续冲了一周后变成了400K。在0流速下确定质谱无污染下重新调谐,但结果为ON PASS。我就用80度的纯开水冲洗脱气机,再用甲醇冲洗脱气机,A、B均用了半小时左右。后来直接LC-MS突然发现了m/z102到了11M,吓我一大头啊!我想来想去是不是哪出问题了,直接把针、针座全换,将喷针直接用异丙醇超声10分钟,配了个异丙醇:乙腈:甲醇=2:1:1,将脱气机A、B冲半小时后直接1mL/min打到MS,m/z102本底有12M。后来1mL/min冲液相部份,第二天来后打MS,发现102只有600K了,不过出现了个m/z425(估计是异丙醇里面的)。换上纯水打到MSm/z102只有8000的本底了,后来我配了个95%甲醇水mL/min打到MS过夜,第二天来后打到MSm/z102只有10K了,将离子源用开水泡洗了5次,调谐了一次就PASS。回头想了想A的工程师用0.5~0.8%乙酸/甲酸水是不是有问题浪费了一周时间,还是我配异丙醇:乙腈:甲醇=2:1:1,效果比0.5~0.8%乙酸/甲酸水好。希望大家发表下见解,一年多的A新手!
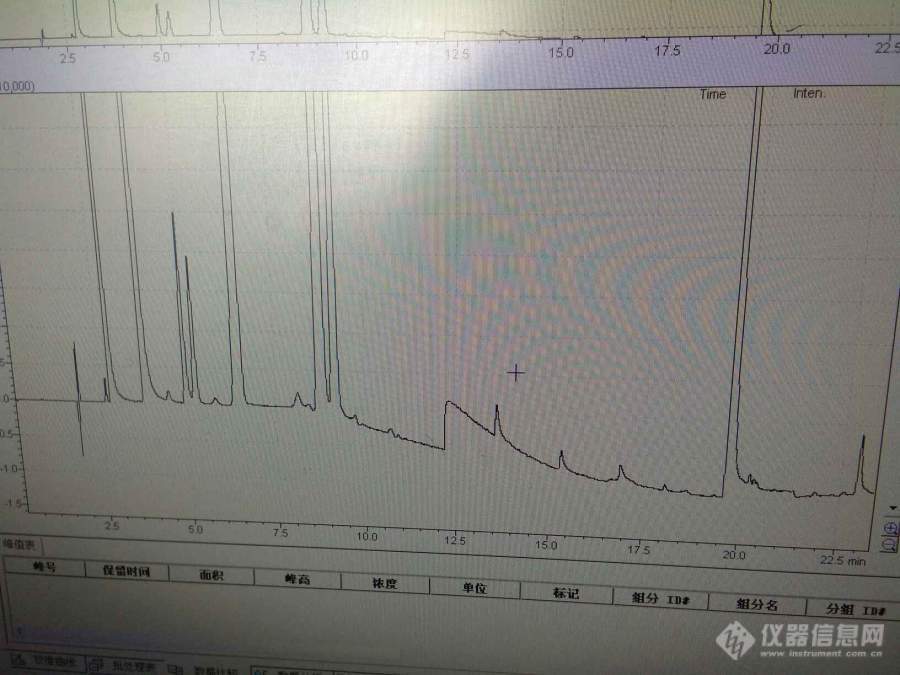
[color=#444444]我想请问一下,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,顶空进样,溶剂为N-甲基吡咯烷酮,柱子DB-624,溶液里面成分有甲醇,乙醇,乙腈,二氯甲烷,正己烷,乙酸乙酯,四氢呋喃,三乙胺,吡啶,DMF,目前三乙胺峰判断不出来,也不确定到底有没有出来峰,图谱已传上,请教一下[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906141009051727_9546_1752329_3.jpg!w690x517.jpg[/img][/color]
做氨基酸用的药品三乙胺,醋酸钠方法上说需要优级纯,可是优级纯的很难买到,用分析纯的可以不?
三乙胺怎么除水呢,我要求不高,查资料说不能用硫酸镁,可以用氯化钙吗







 我要推广仪器
我要推广仪器
 下载APP
下载APP