做了份马来酸氯苯那敏原料药,参照中国药典2010年二部,附上图,发现3400波数左右,出现异常,其他部分还好,不明白原因,有高手援助下!谢谢!
马来酸氯苯那敏在流动相中,很容易分成两个峰:马来酸峰和氯苯那敏峰。在含量计算中,是按氯苯那敏峰计算的。 可是,在有关物质的计算中,主峰也是按氯苯那敏峰计吗?还是按马来酸峰和氯苯那敏峰之和来计算呢?? 如果有关物质计算,主峰只按氯苯那敏峰计算(药典规定,杂质除马来酸峰外),那是否就没有必要积分马来酸峰了??
马来酸氯苯那敏在流动相中,很容易分成两个峰:马来酸峰和氯苯那敏峰。在含量计算中,是按氯苯那敏峰计算的。可是,在有关物质的计算中,主峰也是按氯苯那敏峰计吗?还是按马来酸峰和氯苯那敏峰之和来计算呢??如果有关物质计算,主峰只按氯苯那敏峰计算(药典规定,杂质除马来酸峰外),那是否就没有必要积分马来酸峰了??
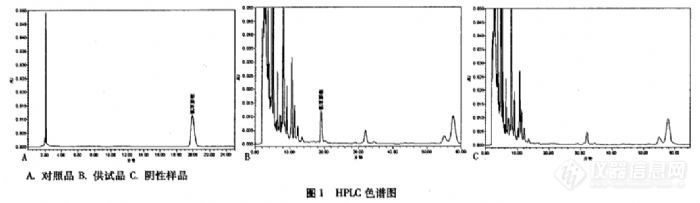
http://simg.instrument.com.cn/bbs/images/brow/emyc1007.gif这是我们实验室几年前做的,拿来参加原创大赛,支持化学药分析版。HPLC法测定小儿氨酚黄那敏片马来酸氯苯那敏含量【处方】 马来酸氯苯那敏 0.5g 对乙酰氨基酚 125g 人工牛黄 5g共制成 1000片1.对照品与供试品马来酸氯苯那敏对照品(中国药品生物制品检定所,批号100047-200305)对乙酰氨基酚对照品(中国药品生物制品检定所,批号100018-200408)小儿氨酚黄那敏片(本公司,批号:20060201、20060801、20060802)小儿氨酚黄那敏片阴性样品(不含马来酸氯苯那敏和对乙酰氨基酚、本公司)2.马来酸氯苯那敏含量测定2.1仪器与试剂2.1.1仪器:岛津LC-10A高效液相色谱仪2.1.2试剂:甲醇、乙腈为色谱纯,磷酸二氢钾、三乙胺、磷酸为分析纯,水为超纯水。2.2测定方法【含量测定】照高效液相色谱法(中国药典2005年版二部附录V D)测定色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;磷酸盐缓冲液(分别取乙腈250ml与0.02mol/L磷酸二氢钾溶液250ml加十二烷基磺酸钠0.68g,振摇使溶解,用磷酸调pH至3.5)-甲醇(10:9)作为流动相,检测波长为215nm,理论塔板数按马来酸氯苯那敏峰计算应不低于3000,马来酸氯苯那敏峰与其他峰的分离度应符合规定。对照品溶液的制备 取马来酸氯苯那敏对照品约20mg,精密称定,置50ml容量瓶中,加甲醇-0.5%醋酸(1:1)混合液适量,振摇使溶解,加上述混合液稀释至刻度,摇匀,精密量取5ml[/fon
有哪位前辈做过马来酸氯苯那敏的残留溶剂,想请教一下,前辈们做的色谱条件和方法,我按照2015版药典做的,吡啶峰出不来
[size=4]碰到很多高效液相色谱测定马来酸氯苯那敏的情况,有时马来酸分成两个峰(马来酸和氯苯那敏峰),有时又不分离,就出一个峰,因为之前没有将出峰情况及条件收集起来,网上也没有查询到马来酸氯苯那敏在各种溶液中的稳定性(酸性,碱性,中性溶液),现在又碰到要测定该项目,且要做方法学验证,希望能在这又一些有用的收获。主要想知道:1.在各种溶液中的稳定性 2.在什么情况下会分离成两个峰 3.分离成两个峰后怎么计算含量 4.分离后的峰计算的含量是否可信[/size]谢谢
有人做过10版药典的马来酸氯苯那敏溶剂残留吗?我觉得药典的标准有点问题,做过的人一起交流交流,介绍介绍经验呗!
作者:刘伟林 广西壮族自治区食品药品检验所监督抽样室,广西南宁市530021摘 要:目的建立高效液相色谱法测定苍鹅鼻炎胶囊中马来酸氯苯那敏的含量的方法。方法采用DiamonsilC18(250mm×4.6mm,5μm)柱:乙腈-0.2%十二烷基硫酸钠溶液-磷酸(50∶50∶0.1)为流动相;流速:1.0ml/min;检测波长为225nm。结果马来酸氯苯那敏线性范围为0.1238~0.2890μg(r=0.9999),平均回收率为99.3%,RSD=0.4%(n=6)。结论该方法简便,准确,可控制苍鹅鼻炎胶囊中马来酸氯苯那敏的含量。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207161209_377839_2379123_3.jpg
有哪位朋友做过马来酸氯苯那敏残留溶剂?烦请传一份对照溶液的气相色谱图看看。想知道各溶剂的出峰顺序及保留时间(我们用的是SE-54柱)。对药典描述的程序升温,不太明白终温保持10分钟,是检测什么成份的。我们这里做的样品及对照在24~32分钟时间段内峰都不成形。http://ng1.17img.cn/bbsfiles/images/2012/08/201208171758_384329_1878194_3.jpg
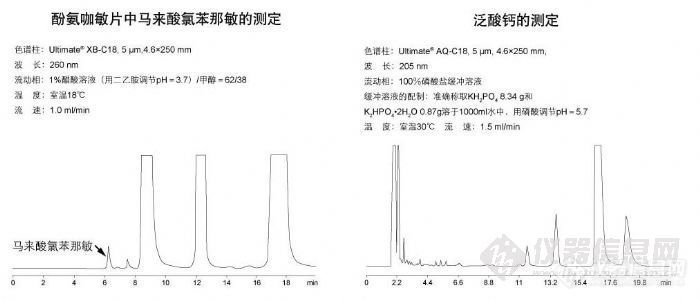
酚氨咖敏片中马来酸氯苯那敏的测定和泛酸钙的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021853_180186_1896702_3.jpg
马来酸氯苯那敏原料检查项下:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定有关物质[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定四氢呋喃、二氧六环、吡啶、甲苯残留溶剂测定缺少:二乙烯基-乙基乙烯苯型高分子小球固定相及与之配套的填充柱诸位大哥,谁知道以上提到的,全部购买需要多少钱?有谁做过?怎么做的呢?
有谁用2010版药典中液相条件做过马来酸氯苯那敏有关物质吗?照药典那个梯度我没做出来,而且大约20分钟的那个峰(第一次做,还不敢确定那个峰是不是氯苯那敏)拖尾很严重。不知道这个实验对柱子的要求高不高,我用的是一根ODSC18。另外想问一下,单位刚买了一台液相,做验证,走了基线,请问如果根据走的基线图来判断漂移和噪音是否合格,有标准吗?请指导一下,谢谢
有没有那位大神有马来酸氯苯那敏[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]残留的图谱看看啊,自己的分离度不太好,不好定位峰
看到马来酸氯苯那敏这个东西,我是觉得既熟悉又陌生,为何这么说呢?说熟悉,是因为在论坛上见过一些版友发帖讨论,说陌生,是自己根本没有亲身接触过,也不知道版友讨论的检测问题是否存在。总结这些讨论问题,大致是说马来酸氯苯那敏会出现两个峰,马来酸氯苯那敏会分解为马来酸和氯苯那敏,在色谱柱上表现为这2个组分。还没做这个工作,上论坛看到这些帖子,都有点让我打颤,如何真是两个峰,让我改怎么办呀。既来之则安之,只好先照药典的方法做吧。可怜的是我现在已经忙得后脚粘前脚了,根本就没有多少时间研究这个家伙,所以检测这个组分,我的检测过程是否正确,我也还说不上,仅以我的处理方法,分享给大家参考,有经验的大侠还希望多提意见和问题呀。色谱柱:UltimateTM液相色谱柱(XB-C18,5um,4.6*250mm)检测波长:264nm流动相:甲醇(含0.5%三乙胺,【注:药典为1%】)+0.05mol/L磷酸二氢钾(用磷酸调节PH=3.0,【注:药典还含0.005mol/L的庚烷磺酸钠,这里未添加】),流速:1.0mL/min对照品溶液的制备和样品溶液的制备,都是按照2010年药典上的方法处理,下图就是我进对照品溶液得到的色谱图,看到这个图,我也是很纳闷,溶剂峰有这样的么?http://ng1.17img.cn/bbsfiles/images/2011/12/201112131311_337805_1608710_3.gif主要的目标峰出现了,后面也没有其他峰,就拿这个漂亮的峰当它了吧,先进个样品再说,样品可是处理了很辛苦来的呀(样品处理过程有点复杂),别浪费了,先瞧瞧样品里面是否有货。下图就是样品的色谱图http://ng1.17img.cn/bbsfiles/images/2011/12/201112131311_337806_1608710_3.gif把样品色谱图和对照品色谱图重叠后显示比较下,http://ng1.17img.cn/bbsfiles/images/2011/12/201112131311_337807_1608710_3.gif比较后,发现开始这个倒峰竟然能完全吻合,难道真是传说中的溶剂峰?按理论分析,定容液是流动相,不可能有溶剂峰出现才对呀?没有其他辙,再进一个空白试一试吧?空白色谱图,无法与对照品和样品图吻合http://ng1.17img.cn/bbsfiles/images/2011/12/201112131312_337808_1608710_3.gif总结:1.没有出现版友说的马来酸氯苯那敏是2个峰的情况。2.我无法确定前面这个倒峰是怎么来的。3.按照后面那个峰来计算样品结果,是基本一致的。 有做过的大虾,你们都遇到了什么问题呢?
我公司一中西药复方制剂中需测定马来酸氯苯那敏含量,采用HPLC法,条件:以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.05mol/l磷酸二氢钾(40:60)(用磷酸调pH值至3.0)为流动相;检测波长为260 nm。限度要求为标示量的90.0%~110.0%。我们在生产时投料量按120%,可实际测定结果老是偏低,为90%左右。其峰型较差,主要是拖尾严重。请问那位老师能分析一下原因,并能提出改进方法,在此不甚感激!!!如方便请回leehb606@sina.com
HPLC法测小儿氨酚黄那敏颗粒中马来酸氯苯那敏的含量。本人查阅了一些文献,各篇文章中均不一样。①大连依利特C18(250mm*4.6mm,5um);流动相是甲醇-0.5mol/l磷酸二氢钠-三乙胺(150:850:0.25),用磷酸调节pH至2.3;检测波长为215nm②Eclipse*DB-C18(250mm*4.6mm,5um);流动相是甲醇-0.05mol/l磷酸二氢钾-三乙胺(10:90:0.02),用磷酸调节pH至3.4;检测波长为215nm③Shim-pack VP-ODS(150mm*4.6mm,5um);流动相是甲醇-0.05mol/l磷酸二氢钠-三乙胺(19:81:0.025),用磷酸调节pH至2.2;检测波长为215nm④Kromasil C18(250mm*4.6mm,5um);流动相是乙腈-0.3%十二烷基硫酸钠溶液-磷酸(60:40:0.02),用三乙胺调pH至3.5;检测波长为254nm有没有谁能帮我分析一下?我们现在有大连依利特和美国天地的柱子,岛津液相色谱仪。谢谢!!!
测定康乐鼻炎片中马来酸氯苯那敏的含量,结果发现偏高,更换操作人员和检验仪器,以及色谱柱,结果还是偏高?请问怎么回事呢?
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
以3%[苯基(50%)甲基聚硅氧烷]作为固定液,白色硅藻土为担体,玻璃柱:1.2m,柱温190℃ 检测器250℃,进样器250℃氢气35ml/min 氮气62ml/min (因为样品溶液拖尾严重所以调大氮气流速)空气300ml/min 进样1微升样品溶液:称取样品约400mg 用二氯甲烷稀释至10ml对照溶液:移取样品溶液1ml至100ml容量瓶,用二氯甲烷定容至100ml.要求:记录色谱图至主成分峰保留时间的2倍。供试品溶液色谱图中有如有杂质峰,各杂质峰面积的和不得大于对照溶液主峰面积。图做出来,样品溶液主峰保留时间约在11分钟峰面积很大拖尾严重,到了30分钟还没完全走完,对照液主峰约在13分钟,峰面积极小(比样品主峰面积的1%小的多,几乎检测不出)。请高手指导下,哪里出了问题??这种情况一针是不是要走到样品溶液主峰完全走完时间的2倍?
药典中规定是用二乙烯基-乙基乙烯苯型高分子小球作为固定相。我想问那相当于我们毛细管柱的什么极性柱呢?
如题,我用的是5%苯基-95%聚硅氧烷毛细柱,升温条件按药典上的,但对照中只有四氢呋喃有峰,另外的三个是二氧六环,吡啶,甲苯都不出峰,是为什么呢,机器各方面正常的,或是换什么条件好,有哪位有经验的帮忙下
四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。问题一:柱温190℃根本无法分离,甲醇峰还没走完 其他峰都出来了。是不是柱子太短了?用柱温190℃做出了的柱子长度是多少?问题二:3年前做过用程序升温,分离还可以,现在用同样的柱子同样条件做分离度不行了,柱子用了很久分离效果为什么会变差?那怎么办?问题三:查资料有报道说苯的浓度要大十倍。大十倍会好些吗?
HPLC测定酚氨咖敏片含量时有几个峰
http://simg.instrument.com.cn/bbs/images/default/emyc1007.gif支持分坛团队和化学药分析版。小儿氨酚黄那敏片原质量标准控制崩解时限,按标准提高要求,需进行溶出度研究。因此我们对小儿氨酚黄那敏片的对乙酰氨基酚溶出度进行了研究。【处方】马来酸氯苯那敏 0.5g对乙酰氨基酚 125g人工牛黄 5g共制成 1000片供试品来源马来酸氯苯那敏对照品(中国药品生物制品检定所,批号100047-200305)对乙酰氨基酚对照品(中国药品生物制品检定所,批号100018-200408)小儿氨酚黄那敏片(本公司,批号:20060201、20060801、20060802)小儿氨酚黄那敏片阴性样品(不含马来酸氯苯那敏和对乙酰氨基酚、本公司)小儿氨酚黄敏片(某公司上市产品,批号:0511141)对乙酰氨基酚溶出曲线的测定溶出曲线应与体内过程一致。初步研究为简便起见,取上市产品同法测定,对比溶出曲线,看是否有相关性。溶出介质的选择溶出介质首选水,对乙酰氨基酚在水中略溶,即1g能在溶剂30~不到100ml中溶解。本品每片含对乙酰氨基酚0.125g,不考虑其它成分干扰的话,0.125g对乙酰氨基酚应该能在1000ml的水中完全溶解。溶出方法的选择一般认为,桨法50转相当于篮法100转。本研究先从缓和的条件篮法50转开始。溶出度曲线测定方法参照中国药典2005年版对乙酰氨基酚片溶出度项下的有关规定,拟定为“取本品,照溶出度测定法(附录X C第一法),以水1000ml为溶出介质,转速为每分钟50转,依法操作,经5分钟、15分钟、30分钟、45分钟时,取溶液10ml,滤过,精密量取续滤液3ml,加0.04%氢氧化钠溶液稀释至50ml,摇匀,照紫外-可见分光光度法(附录IV A),在[
[em06] 四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。我让色谱公司按这个要求做了不锈钢柱子(柱填料:401有机载体(二乙烯基苯/乙基乙烯苯共聚体)60-80目),可是不出峰,后来把柱寄回去了,现在又寄给我的柱子(柱填料:10%PEG-20M CHROMOSORB PAW-DMCS 80-100目),峰是有了,可是分不开,我做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的氮气4圈,空气4.2圈,氢气4.5圈,后我又把氮气开到3圈,还是这个样子.是怎么回事呢,请高手赐教.谢谢!!!
马来酸二甲酯和苯甲醇这两者的化学分子式是什么在高温下,又会产生什么?
最好加上前处理!谢谢~谢谢`!
[em09508]有那位大虾可以告诉我,分离对乙酰氨基酚(Paracetamol),盐酸伪麻黄碱(Pseudoephedrine Hydrochloride),氢溴酸右美沙芬(Dextromethorphan hydrobromide),马来酸氯苯那敏(hlorphenamine)这四种物质,应该用什么类型的液相色谱柱!!

10,抽取5个版友);中奖名单:zengzhengce163(注册ID:zengzhengce163)大川之子,纵横四海(注册ID:chuangu120)莫名其妙(注册ID:moyueqiu)WUYUWUQIU(注册ID:wulin321)千层峰(注册ID:jxyan)http://ng1.17img.cn/bbsfiles/images/2016/06/201606241516_598031_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/06/201606241517_598032_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。======================================================================= 止咳糖浆方法:HPLC基质:动物提取物应用编号:101156化合物:氢溴酸右美沙芬;麻黄碱;马来酸氯苯那敏固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil 5μm C18(2), 250 x 4.6mm色谱条件:流动相:乙腈/缓冲溶液 流速:1.0 mL/min 温度:30 oC 检测器:UV 220 nm文章出处:AN: D1119关键字:止咳糖浆,HPLC,Diamonsil C18(2),钻石二代,氢溴酸右美沙芬;麻黄碱;马来酸氯苯那敏谱图:http://www.dikma.com.cn/Public/Uploads/images/D1119%20copy.png图例:1. 氢溴酸右美沙芬;2. 麻黄碱;3. 马来酸氯苯那敏
马来酸二甲酯和苯甲醇在反应时颜色变黄,不知道是什么原因阿,有专家吗







 我要推广仪器
我要推广仪器
 下载APP
下载APP