脂肪酸气相色谱分析的故事
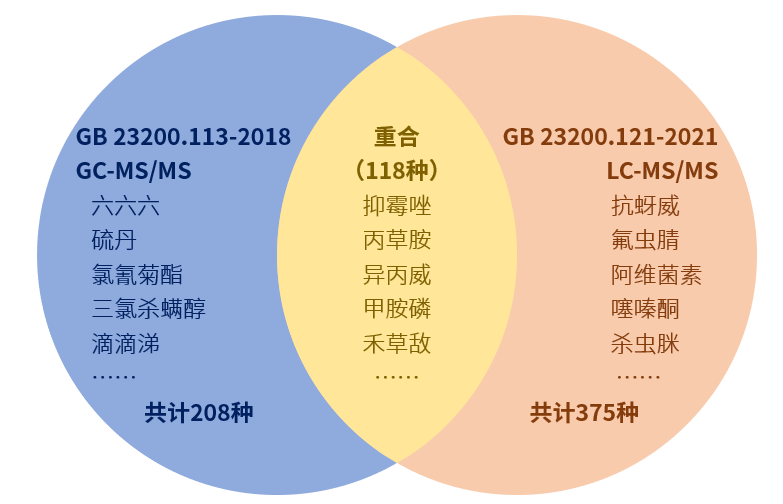
编者注:傅若农教授生于1930年,1953年毕业于北京大学化学系,而后一直在北京理工大学(原北京工业学院)从事教学与科研工作。1958年,傅若农教授开始带领学生初步进入吸附柱色谱和气相色谱的探索 1966到1976年文化大革命的后期,傅若农教授在干校劳动的间隙,系统地阅读并翻译了两本气相色谱启蒙书,从此进入其后半生一直从事的事业——色谱研究。傅若农教授是我国老一辈色谱研究专家,见证了我国气相色谱研究的发展,为我国培养了众多色谱研究人才。 第一讲:傅若农讲述气相色谱技术发展历史及趋势第二讲:傅若农:从三家公司GC产品更迭看气相技术发展第三讲:傅若农:从国产气相产品看国内气相发展脉络及现状第四讲:傅若农:气相色谱固定液的前世今生第五讲:傅若农:气-固色谱的魅力第六讲:傅若农:PLOT气相色谱柱的诱惑力第七讲:傅若农:酒驾判官——顶空气相色谱的前世今生第八讲:傅若农:一扫而光——吹扫捕集-气相色谱的发展第九讲:傅若农:凌空一瞥洞察一切——神通广大的固相微萃取(SPME)第十讲:傅若农:悬“珠”济世——单液滴微萃取(SDME)的妙用第十一讲:傅若农:扭转乾坤——神奇的反应顶空气相色谱分析第十二讲:擒魔序曲——脂质组学研究中的样品处理第十三讲:离子液体柱——脂质组学中分离脂肪酸的气相色谱柱 上一讲我们主要介绍了在脂质组学中对脂肪酸的分析所用的离子液体毛细管色谱柱,但是用气相色谱分析脂肪酸源远流长,有许多故事,了解一些过去的故事对现在的发展理解有好处,温故才可以知新。 先讲一下脂质组学中常常要研究的血浆分析,其中一个重要的项目是分析其中的脂肪酸,下面一个例子,概要介绍了血浆中脂肪酸的主要成分: “虽然游离脂肪酸只占血浆中脂肪酸的一小部分,但它代表一类高度代谢活性的脂质,脂肪组织是血浆游离脂肪酸的主要来源,其分布与食物的脂肪酸组成密切相关。在正常情况下从脂肪组织中释放脂肪酸与组织对能量的需要紧密相连。但是当代谢失调时,这种平衡被打乱,导致脂解增加,会释放出多于组织所需要脂肪酸的量。健康人经过一夜禁食后血浆中含有214 nmol/ml游离脂肪酸,油酸(18:1)的含量最高,其次是棕榈酸(16:0)和硬脂酸(18:0),这三种酸占全部游离脂肪酸的78%。亚油酸(18:2)和花生四酸(20:4) 是主要的多不饱和脂肪酸(约占8%)。但是有营养作用的α-亚麻酸(18:3ω-3),二十碳五烯酸(20:5, EPA)和二十二碳六烯酸(22:6, DHA)也占有一定比例,约为全部游离脂肪酸的1%。”1 脂肪酸气相色谱分析的历史故事 气相色谱被认为是分析复杂混合物中脂肪酸的可靠方法,这一方法可追述到上世纪50年代,气相色谱的出现于脂肪酸的分析有密切的关系,1952年气相色谱发明人A. T. James 和 A. J. P. Martin就用最为原始的自制气相色谱仪分析小分子脂肪酸(Biochem J,1952,50:679),他们首次阐明气-液分配气相色谱的原理,设计了自动滴定检测脂肪酸的气相色谱仪。实验过程中使用的色谱柱为玻璃柱,其内径为4mm,长度为5英尺,固定相是把DC 550硅油涂渍在硅藻土Celite 545上。分离小分子脂肪酸的色谱如图1所示。 图1 用自动滴定计气相色谱仪分析小分子脂肪酸的色谱图 分离从乙酸到戊酸的色谱如图2所示:图 2 分离从乙酸到戊酸的色谱 此后分析脂肪酸的一个重大进步是把脂肪酸进行甲酯化,1956年James和Martin使用气体密度检测器,并把脂肪酸进行甲酯化,使用阿皮松类高温润滑脂作固定相,可以分离分子量大的脂肪酸。图3 是分离C5-C13直链和支链脂肪酸甲酯的色谱图。图 3 用高沸点润滑脂分离C5-C13直链和支链脂肪酸甲酯的色谱图色谱柱:在硅藻土载体上涂渍高沸点润滑脂;柱温:197℃;载气:氮气 14.1mL/min 色谱峰: (1) 空气, (2) n-戊酸甲酯,(3) n-己酸甲酯, (4) 4-甲基己酸甲酯,(5) 6-甲基庚酸甲酯, (6) n-辛酸甲酯, (7) 6-甲基辛酸甲酯, (8) n-壬酸甲酯,(9) 8-甲基壬酸酯, (10) n-癸酸酯, (11) 8-甲基癸酸酯, (12) 10-甲基十一酸酯 ,(13) n-十二酸酯, (14) 10-甲基十二酸酯2 脂肪酸气相色谱分析的发展 脂肪酸的气相色谱分析由于它的极性和挥发性不好而带来麻烦,所以首先要把它的极性羰基转化成易于挥发的非极性衍生物。有多种烷基化试剂可以进行羰基的衍生化,使用最多的是进行甲基化,特别是使用氢火焰离子化监测器(FID)气相色谱时,尤为方便普及。但是使用FID也有一些不足之处。绝对的定量要依靠内标物的信号强度,经常使用的内标物是十七酸(而不是使用化学和物理性质与所测定脂肪酸相近的同位素标记脂肪酸混合物作内标)。人类体内不能合成奇数碳链的脂肪酸(包括碳17酸),但是人们可以通过食物摄取它们,它们存在于血液的血浆中,增加内标物十七酸的量,从而扰乱定量分析。 进一步讲,FID不能提供分子质量或其他结构特征信息,以便区分不同的脂肪酸,所以色谱和FID只是解决把所有要研究的脂肪酸分子完全分离开,用质谱解决脂肪酸的结构信息。大家应该知道使用电子轰击电离脂肪酸分子很容易被打成碎片,通过这些碎片可以进行脂肪酸的结构分析,但是灵敏度受到限制。弱电离技术比如负化学电离(NCI)可以改善检测限。使用卤代衍生化试剂可以进一步提高检测灵敏度,这种试剂增加了电子亲和力,可改善NCI-MS的灵敏度。Kawahara 使用五氟基苄(PFB) 作衍生化试剂来衍生化有机羧酸,这样的含氟衍生物电子很容易被俘获。此后这一方法扩展到脂肪酸的衍生化为脂肪酸酯,与脂肪酸甲酯相比,它很容易被NCI-MS检测。所以使用五氟基苄进行衍生化有利于提高检测灵敏度。许多研究者使用PFB做衍生化试剂进行脂质组学中的脂肪酸分析,例如Quehenberger等就是用这一方法分析巨噬细胞中的各种脂肪酸(Prostaglandins, Leukotrienesand Essential Fatty Acids,2008,79:123–129)。下图4 是分析巨噬细胞中的各种脂肪酸的色谱图。图 4 巨噬细胞中的各种脂肪酸的色谱图图中色谱峰的脂肪酸如下:(1)12:0 (2)14:0 (3)15:0 (4)16:1 (5)16:0 (6)17:1 (7)17:0 (8) a18:3 (9) 18:4 (10) g18:3 (11)18:2 (12)18:1 (13)18:0 (14)20:4 (15)20:5 (16)11,14,17–20:3 (17)bishomo-20:3 (18)20:2 (19)5,8,11–20:3 (20)20:0 (21)22:6 (22)22:4 (23)22:5 (24)22:2 (25)22:3 (26)22:1 (27)22:0 (28) 23:0 (29)24:1 (30)24:0 3 国内外进行气相色谱分析脂肪酸的一些例证 为了进一步了解进行气相色谱分析脂肪酸的具体情况,下面表1列出近50例分析各种样品中脂肪酸的色谱柱和分离对象。表2列出国外文献中分析人体组织中脂肪酸的例证。表 1 国内气相色谱分析脂肪酸的色谱柱和分析对象 表 2 国外文献中有关分析人体组织中脂肪酸的衍生化方法和所用色谱柱4 脂肪酸气相色谱分析所用色谱柱 从已发表的文献看分析整体脂肪酸需用非极性的聚硅氧烷毛细管色谱柱,如聚二甲基硅氧烷,分离多不饱和脂肪酸需用极性强的色谱柱,如OV-275,OV-275(这是聚硅氧烷固定相中极性最强的色谱柱)和CP-Sil 88(HP-88)。 据安捷伦公司一份研究报告(5989-3760 EN),他们对最重要的一些脂肪酸(甲酯)(见表3)进行研究,研究总结认为:聚乙二醇柱对不太复杂的样品可以得到很好的分离 而中等极性的氰丙基聚硅氧烷柱(DB 23)对复杂的 FAMEs 样品可以得到很好的分离,对一些顺反异构体也可以得到分离 要使顺反异构体分离的更好,就要使用更高极性的 HP-88 氰丙基色谱柱。表3 重要的一些脂肪酸 三种主要色谱柱分离脂肪酸的特点如下: 使用DB-Wax柱,DB-23 柱和HP-88 柱上分离37种脂肪酸混合物的色谱见图5-图7.图 5 FAMEs在30 m 0.25 mm ID, 0.25 μm DB-Wax 色谱柱上的色谱图 6 FAMEs混合物在 60 m 0.25 mm ID, 0.15 μm DB-23 柱上的色谱图 7 FAMEs 混合物 在 100 m 0.25 mm ID, 0.2 μm HP-88 柱上 的色谱 其中HP-88 柱的极性最强,是含88%氰丙基甲基聚硅氧烷,其结构如下图8:图8 HP-88 的分子结构 HP-88 对一些异构体的分离能力由于DB-23如下图9所示 图 8 HP-88和HP-23分离能力的差别 (此图来自Walter Jennings博士2008年在北京大学作报告时的ppt文稿) 吴惠勤等使用P-88毛细管色谱柱分离了39种脂肪酸得到的质谱基峰离子和特征离子如表4中的数据。表4 39种脂肪酸在HP-88毛细管色谱柱上出峰次序( 吴惠勤等,分析化学,2007,35(7):998-1003)
























 我要推广仪器
我要推广仪器
 下载APP
下载APP