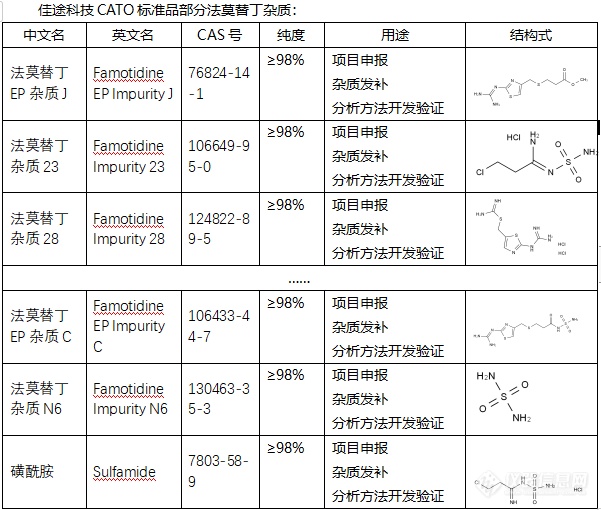
[font=宋体]◇关于法莫替丁杂质[/font][font=Helvetica][color=#333333] 法莫替丁,[/color][/font][font=宋体][color=#333333]英文名是[/color][/font][font=Helvetica][color=#333333]Famotidine[/color][/font][font=宋体][color=#333333],[/color][/font][font=Helvetica][color=#333333][font=Helvetica]化学式为[/font]C[/color][/font][font=Helvetica][color=#333333]8[/color][/font][font=Helvetica][color=#333333]H[/color][/font][font=Helvetica][color=#333333]15[/color][/font][font=Helvetica][color=#333333]N[/color][/font][font=Helvetica][color=#333333]7[/color][/font][font=Helvetica][color=#333333]O[/color][/font][font=Helvetica][color=#333333]2[/color][/font][font=Helvetica][color=#333333]S[/color][/font][font=Helvetica][color=#333333]3[/color][/font][font=Helvetica][color=#333333][font=Helvetica],是一种组胺[/font]H[/color][/font][font=Helvetica][color=#333333]2[/color][/font][font=Helvetica][color=#333333]受体拮抗剂[/color][/font][font=宋体][color=#333333]的[/color][/font][font=Helvetica][color=#333333]有机化合物[/color][/font][font=Helvetica][color=#333333],[/color][/font][font=宋体][color=#333333]在临床上[/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]法莫替丁是一种重要的药物,广泛用于治疗胃酸过多[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe],[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]还有相关[/back][/color][/font][font=Helvetica][color=#333333]胃[/color][/font][font=宋体][color=#333333]疾病以[/color][/font][font=Helvetica][color=#333333]及十二指肠溃疡、反流性食管炎、上消化道出血[/color][/font][font=宋体][color=#333333]等。它的原理机制是[/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]通过阻[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]止并且切断[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]胃黏膜壁细胞中的H2受体,[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]达到[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]抑制胃酸的分泌[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]的效果[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]。法莫替丁还可以用于预防应激性溃疡的发生,例如在重大手术或严重创伤后。[/back][/color][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的法莫替丁杂质用途主要是用于分析化学物质和质量控制的化学物质。[/font][/font][img=,601,510]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052224404125_7305_6381607_3.png!w601x510.jpg[/img][font=宋体][font=宋体] 广州佳途科技股份有限公司,[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供法莫替丁全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,严格的控制质量,通过全面的检测,[/font][font=微软雅黑][color=#444444]高效的沟通,专业的服务,完善的售后[/color][/font][font=微软雅黑][color=#444444],[/color][/font][font=宋体]所有产品均能现货供应[/font][font=宋体]。[/font]
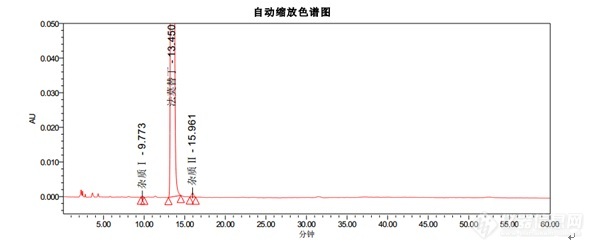
[align=center][b]法莫替丁颗粒系统适用性试验-2015中国药典 [/b][/align][align=center] [/align]色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相:醋酸盐缓冲溶液(取醋酸钠 13.6g,溶于900ML水中,用冰醋酸调节pH至6.0±0.1,加水至1000ML):乙腈=93:7流速:1.5ml/min柱温:35℃波长:270nm进样量:20[color=#333333]μL[/color][color=#333333][img=,596,251]https://ng1.17img.cn/bbsfiles/images/2018/12/201812271539396153_2091_2428063_3.jpg!w596x251.jpg[/img][/color]结论:1. 出峰顺序为杂质Ⅰ,法莫替丁,杂质Ⅱ2. 法莫替丁保留时间约为13.4min3. 杂质Ⅰ峰和杂质Ⅱ峰相对于法莫替丁的保留时间约为0.7和1.24. 理论塔板数按照法莫替丁计算不低于5000以上指标均符合中国药典。
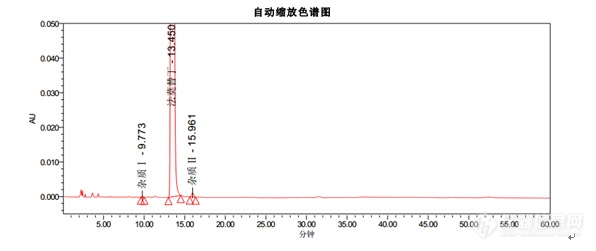
色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相:醋酸盐缓冲溶液(取醋酸钠 13.6g,溶于900ML水中,用冰醋酸调节pH至6.0±0.1,加水至1000ML):乙腈=93:7流速:1.5ml/min柱温:35℃波长:270nm进样量:20μL[align=center][img=,596,251]https://ng1.17img.cn/bbsfiles/images/2018/12/201812101526522191_3600_3232762_3.png!w596x251.jpg[/img][/align][align=left]结论:[/align]1. 出峰顺序为杂质Ⅰ,法莫替丁,杂质Ⅱ2. 法莫替丁保留时间约为13.4min3. 杂质Ⅰ峰和杂质Ⅱ峰相对于法莫替丁的保留时间约为0.7和1.24. 理论塔板数按照法莫替丁计算不低于5000以上指标均符合中国药典。[hr/][align=center]Kromasil品牌[/align]Kromasil是Nouryon旗下高效化学品著名品牌,是全球领先的高性能硅胶基质填料和液相色谱柱生产商。Kromasil高性能多孔球形硅胶基质填料可广泛应用于胰岛素及其类似物、比伐卢定、利拉鲁肽、胸腺法新、达托霉素、EPO等蛋白、多肽及小分子药物等的分离纯化。30年来,Kromasil的经营理念始终是:为制药行业提供以硅胶为基质的、高性价比的、用于医药分离纯化的色谱填料和用于药物分析的液相色谱柱。Kromasil,一以贯之,创新向前。[align=center][img]https://mmbiz.qpic.cn/mmbiz_png/OeFA8HArUwdQyiaia3mAT7HllVGzL6MsslxRXMs2mHMtspgIicoVZic1d5iasgCuC61vnBQBiaC9v88vVZJlTMWwlDMg/640?wx_fmt=png&tp=webp&wxfrom=5&wx_lazy=1&wx_co=1[/img] [/align][align=center] 更[color=#000000]多资料请访问[/color]:[b][color=#007aaa]http://www.kromasil.com/[/color][/b][/align]
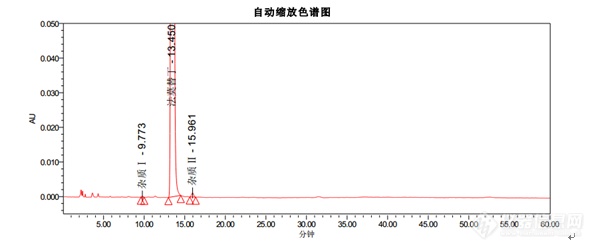
色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相:醋酸盐缓冲溶液(取醋酸钠 13.6g,溶于900ML水中,用冰醋酸调节pH至6.0±0.1,加水至1000ML):乙腈=93:7流速:1.5ml/min柱温:35℃波长:270nm进样量:20μL[align=center][/align][align=center][img=,596,251]https://ng1.17img.cn/bbsfiles/images/2018/12/201812101529374292_9755_2785_3.png!w596x251.jpg[/img][/align]结论:1. 出峰顺序为杂质Ⅰ,法莫替丁,杂质Ⅱ2. 法莫替丁保留时间约为13.4min3. 杂质Ⅰ峰和杂质Ⅱ峰相对于法莫替丁的保留时间约为0.7和1.24. 理论塔板数按照法莫替丁计算不低于5000以上指标均符合中国药典。[hr/][align=center]Kromasil品牌[/align]Kromasil是Nouryon旗下高效化学品著名品牌,是全球领先的高性能硅胶基质填料和液相色谱柱生产商。Kromasil高性能多孔球形硅胶基质填料可广泛应用于胰岛素及其类似物、比伐卢定、利拉鲁肽、胸腺法新、达托霉素、EPO等蛋白、多肽及小分子药物等的分离纯化。30年来,Kromasil的经营理念始终是:为制药行业提供以硅胶为基质的、高性价比的、用于医药分离纯化的色谱填料和用于药物分析的液相色谱柱。Kromasil,一以贯之,创新向前。[align=center][img]https://mmbiz.qpic.cn/mmbiz_png/OeFA8HArUwdQyiaia3mAT7HllVGzL6MsslxRXMs2mHMtspgIicoVZic1d5iasgCuC61vnBQBiaC9v88vVZJlTMWwlDMg/640?wx_fmt=png&tp=webp&wxfrom=5&wx_lazy=1&wx_co=1[/img] [/align][align=center] 更[color=#000000]多资料请访问[/color]:[b][color=#007aaa]http://www.kromasil.com/[/color][/b][/align]
最近REACH指令给出了15种高注物质的清单,其中的丁基锡化合物,我查了很多资料,也没有找到相关的测试标准,仅查到一些文献资料,提出用四乙基硼酸钠进行衍生化,再提取衍生物来进行测试,但具体用什么溶剂提取,怎么做衍生化没有详细的说明,如有知道标准方法的老师,请告知标准号,急需!
急求乙烯中羰基化合物测定的相关标准,多谢!
急求乙烯中羰基化合物测定的相关标准,多谢
安捷伦chemsation的RTL功能一个方法只能锁定一种化合物吗?若能同时锁定多种化合物,是不是对于每种化合物都要在5种不同压力下验证?这样的话岂不是非常繁琐?
请教: 成盐的手性化合物的分析和制备请问如果一个化合物成盐,包括盐酸盐,三氟乙酸盐和有机盐。1. 在手性分析时,该化合物是什么状态在手性柱上分离的?游离态还是成盐的状态?如果化合物是成的有机盐,该有机盐可以由UV检测,那么分析其消旋体时,分析上应该有几根峰?2 . 如果手性液相制备的话,成盐的化合物制备完后是什么状态,游离or成盐?如,一个碱性化合物成盐酸盐,经带有二乙胺的流动相(正己烷:乙醇)制备后还是盐酸盐吗?另外,在手性制备后处理时,二乙胺一般如何除去?旋蒸后还有很多残余~~
http://sioc-journal.cn/Jwk_yjhx/CN/column/item98.shtml2012.10‘有机化合物命名原则’(2010)修订工作 化学名词审定委员会有机化学分委员会决定开展对‘中国化学会’1980年颁发的‘有机化学命名原则’进行修订,为集思广益做好这一工作,修订工作组拟采取边修订,边上网,以征求意见稿的方式分批公告已起草的‘有机化合物命名原则’(2010)。欢迎各界从事和关心有机化学的人士提供宝贵意见,以使尽快高质量地完成这次修订工作,对修订工作的意见请发至信箱:nomencl@sioc.ac.cn现先公告‘有机化合物命名原则’(2010)征求意见稿(2012.10稿)的目录,引言(未全),第一章至第七章和第九章。第八章的草稿和已起草各章进一步的修改稿收到后我们将随时发布。 化学名词审定委员会‘有机化合物命名原则’(2010)修订工作组2012.10 说明和意见征求 下载第一章 有机化合物名称构词概要 下载第二章 有机化合物命名通则 下载第三章 母体氢化物以及由此形成的取代基 下载I 下载II第四章 特性基团(官能(基)团)第五章 命名实施导引 第四章和第五章下载第六章 各类化合物的命名 下载第七章 立体化学 下载第八章 天然产物 (尚未完成)第九章 同位素改变化合物 下载
[size=3][b]绝对构型的确认[/b][/size]希望知道如果一对手性化合物,如果用手性柱拆分开后,在没有文献可以参考的情况下,如果确定他们的绝对构型?
化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。
是不是所有的化合物都能在TIC图上显示?比如4个化合物应该有4个色谱峰?如果只看见2个峰是咋回事呢?
请教大家:我现在在做一个化合物,是枸橼酸钾、枸橼酸钠通过特殊的化学反应生成一种离子化合物。但是现在有个问题,两者反应后不可能全部生成化合物,肯定还有没有反应的物质,如果生成率小于80%那不能满足我的要求了。我现在想知道的是里面有多少“枸橼酸钾钠”化合物,还有他们的分子构成是什么,该用什么方法分析呢?
固定源氯苯类化合物标准中精密度:模拟无组织排放检测点空气采样后测定,是采样还是不采样?直接加标至活性炭中然后解析吗?还是加标后连接空气采样器采样完了再解析?
我想分析含有十二烷基苯酚及其同分异构体的化合物,请问用什么色谱柱好呢 ,有哪位做过相关的分析吗?谢谢大家
顶空可以做强极性化合物吗

【背景介绍】喹宁酸类化合物具有多种具有多种生物活性,如抗组胺、抗氧化、肝保护、抗菌等。而且,其单取代、双取代或三取代化合物在体内化外都显示出良好的抗HIV病毒活性,具有很好的开发前景。【喹定酸类化合物结构】二取代类喹宁酸类化合物是指在R1和R2、R1和R3、或者R2和R3两个位置均有取代的化合物。取代基可能为咖啡酰基(A)、芥子酰基(B)、阿魏酰基(C)等基团的自由组合,即R1、R2、R3可能为A、B、C的排列组合,R4可能为氢也可能为甲基或者乙基等基团。由于R1、R2、R3所处取代位置的化学环境相似,所以在结构鉴定过程中,取代位置容易混淆。http://ng1.17img.cn/bbsfiles/images/2011/10/201110042032_321102_1745326_3.jpg【文章内容】本文通过自己制备的部分单体化合物,利用此类化合物在液相中保留时间的不同,对某植物中存在的部分喹宁酸类化合物(10个:G1~G9)进行指认(涉及保密,本文只透露2个化合物(G4、G7)的相关内容)。对于明确此植物的化学成为具有重要的指导意义。【仪器试剂】水:重蒸水甲醇:色谱纯,天津大茂 乙酸:分析纯,天津大茂 HPLC:Waters 高效液相Waters 996 DAD检测器Waters Model 600 controller液相色谱【色谱条件】 色谱柱:Ultimate XB-C18柱(5μm, 4.6x250mm)流动相:60%甲醇/水(V:V),水含0.01%乙酸流速:1.0ml/min柱温:常温(约25度);检测波长:全波长检测;进样量:10微升;【样品制备】涉及保密,此处略去,见谅。重点在色谱行为。【色谱行为】 某植物样品——总样分析色谱图http://ng1.17img.cn/bbsfiles/images/2011/10/201110062154_321549_1745326_3.jpg指认的G4峰:3,4-二咖啡酰基喹宁酸甲酯http://ng1.17img.cn/bbsfiles/images/2011/10/201110062155_321550_1745326_3.jpg指认的G7峰:3-咖啡酰基-5-芥子酰基喹宁酸甲酯http://ng1.17img.cn/bbsfiles/images/2011/10/201110062156_321551_1745326_3.jpg【结果与讨论】1. 本实验对某植物中所含的喹宁酸类化合物进行了指认(约10个),明确了该植物中含有的化学成分,为以后工作的开展奠定了一定的基础。同时,从色谱图中可见,还有部分化合物由于量小未进行指认,这部分工作将是以后工作的重点;2. G4和G7的色谱图虽然可见一些含量很少的小杂质,但并不影响此次实验分析。由于双取代基团的存在,使得它们的紫外吸收峰并不“干脆”,如果是单取代吸收,将在230nm和320nm处显示两个“干脆尖锐”的紫外吸收峰。这一点,有利于识别单取代和双取代喹宁酸类化合物。3. 另外,不得不反复提出,在进样量不同的时候,通过DAD检测器得出的紫外吸收值有所变化。这提示我们,DAD检测器得出的紫外吸收只应该做为一个参考值,需要引起注意。4. 总样分析色谱图中,由于出峰时间过短及流动相等条件摸索不到位,部分峰形没有完全分离,此部分工作将继续进行。尽管如此,Ultimate XB-C18柱在分析的过程中,表现出了分离度好、柱效高、柱压稳定等优秀性能,让人满意。后续开展的工作,如果涉及指纹图谱类似的相关实验内容,Ultimate XB-C18柱将成为种子选手,绝对首发。
如题,对于一些不稳定的化合物如见光分解,遇空气变坏等等,大家是怎么对它进行定量测定的呢?请教
自今年7月起,欧盟执行2009/425/EC指令,从而正式开始限制对消费产品中特定有机锡化合物的使用。指令2009/425/EC中规定:自2010年7月1日起,欧盟在所有消费品中限制使用三丁基锡和三苯基锡化合物,其限量要求为商品中锡含量的质量百分比浓度小于0.1%,如若检出超标,则该批消费品将遭到退货乃至严厉的召回处罚。 本项指令中关注的有机锡化合物包括三丁基锡、三苯基锡化合物及二丁基锡、二辛基锡化合物,其中前两者的正式开始限制时间为2010年7月1日,而后两者的时间则为2012年1月1日。以上四种有机锡化合物被广泛地应用于消费品中,例如鞋的内底,袜子和运动衣的抗菌整理,聚氨酯泡沫生产过程中的添加剂,PVC生产过程中的稳定剂或硅橡胶生产过程中的催化剂等。据统计,在现实生产过程中,全世界的锡产量中的10%~20%是用于合成有机锡化合物的,由此可见该物质应用的广泛程度。并且有机锡化合物对生物体的危害严重,会引起糖尿病和高血脂病等。 据统计,2010年上半年,宁波口岸出口至欧盟的商品共计62413批次,合15.72亿美元,相比2009年同期,分别提高了27.0%和26.6%,呈现出良好的上升态势,其中主打的拳头产品包括纺织品、玩具产品、食品接触类材料等,这些物品在生产加工过程中都有可能会添加有机锡化合物,如果这些潜在含有有机锡化合物的产品未通过检测贸然输往欧盟,可能会导致大规模的退货乃至召回的后果,这将会严重影响“中国制造”在欧盟的声誉,最终会对正处在逐渐回暖过程中的中欧贸易造成不可预计的恶性后果。 为此,检验检疫部门提醒:第一,输欧消费类产品的生产企业要加强原辅材料和生产过程的管理,要求原辅材料供应商提供不含有机锡化合物的检测报告,同时积极改进加工工艺,确保整个生产过程不添加有机锡化合物;第二,相关企业应积极通过与政府职能部门的配合,获取更多的有毒有害物质检测技术和检测标准知识,稳固企业技术储备工作;第三,检验检疫部门应加大对相关商品的有机锡化合物的抽样检测工作力度,以保证起到切实有效的监管作用;此外,检验检疫部门还可以考虑在国际层面上加强与欧盟在有毒有害物质管理方面的信息交换和有效配合,掌握国外有毒有害物质最新标准的发展趋势,以利于企业进行各项技术创新和管理变革。
衍生反应是否会影响手性化合物的光学纯度如题,有没有哪个高手做过这方面的课题研究??
赛默飞[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]TSQ. QUANTIS化合物参数优化的具体操作规程是怎样的
用4-氨基安替比林测大气中的酚类化合物,做标线,线性相关系数才1个久,截距是个负数。。做了好几遍了,线性都不好,截距也不接近于零,我是不是哪些地方没有注意呢,做标线的过程中需要注意些什么吗?
自19世纪末以来,稀有气体元素不能生成热力学稳定化合物的结论给科学家人为地划定了一个禁区,致使绝大多数化学家不愿再涉猎这一被认为是荒凉贫瘠的不毛之地,关于稀有气体化学性质的研究被忽略了。尽管如此,仍有少数化学家试图合成稀有气体化合物。1932年,前苏联的阿因托波夫(A.R.Antropoff)曾报道,他在液体空气冷却器内,用放电法使氪与氯、溴反应,制得了较氯易挥发的暗红色物质,并认为是氪的卤化物。但当有人采用他的方法重复实验时却未获成功。阿因托波夫就此否定了自己的报道,认为所谓氪的卤化物实际上是氧化氮和卤化氢,并非氪的卤化物。1933年,美国著名化学家鲍林(L.Pauling)通过对离子半径的计算,曾预言可以制得六氟化氙(XeF6)、六氟化氪(KrF6)、氙酸及其盐。扬斯特(D.M.Younst)受阿因托波夫的第一个报道和鲍林预言的启发,用紫外线照射和放电法试图合成氟化氙和氯化氙,均未成功。他在放电法合成氟化氙的实验中将氟和氙按一定比例混合后,在铜电极间施以30000伏的电压,进行火花放电,但未能检验出氟化氙的生成。扬斯特由于对传统观念心有余悸,没有坚持继续进行实验,使一个极有希望的方法半途而废。一系列的失败,致使在以后的30多年中很少有人再涉足这一领域。令人遗憾的是,到了1961年,鲍林也否定了自己原来的预言,认为“氙在化学上是完全不反应的,它无论如何都不能生成通常含有共价键或离子键化合物的能力”。
请问谁有4氨基安替比林分光光度法测定的酚类物质的标准曲线啊,是按照国标号HJ/T32-1999做的,固定污染源中酚类化合物的测定,我做了一个不知道哪里出了问题乱糟糟的,谁有的话给我一个看一下啊,找找区别,万分感谢啊
极性化合物完美分离保留 Atlantis色谱柱 给极性化合物和非极性化合物的保留提供了完美的平衡下载Atlantis色谱柱介绍资料(PDF) Atlantis 色谱柱使用高纯度硅胶及双键键合 C18 技术,并对填料的孔径大小、端基封口以及 C18 的键合密度进行优化,从而使 Atlantis 色谱柱具有: 对极性化合物保留能力强,在水流动相中性能稳定,低 pH 条件下色谱柱寿命长,与质谱兼容,色谱峰形优异,重现性好等优点。Atlantis 色谱柱目前提供3um和5um两种粒度填料,键合相类型则有dC18和HILIC(亲水交互作用)两种类型。Atlantis 资料目录:极性化合物的保留ATLANTI dC18 反相HPLC分析的理想选择极性化合物的保留增强色谱柱的长寿命和低pH稳定性为使用水溶液流动相优化的填料即使没有内嵌极性官能团也与水溶液兼容完全端基封口色谱柱的优势极性和非极性化合物保留的最佳平衡ATLANTI dC18 色谱柱在肽谱方面的应用ATLANTI dC18快速分析柱优异的重现性轻松放大、更长、可预计的色谱柱寿命保留和高上样量的优化亲水相互作用色谱柱(HILIC)ATLANTIS HILIC 硅胶色谱柱适合于在反相色谱柱上不能保留的化合物的分离HILIC能够提高ESI-MS的灵敏度简化样品制备过程Atlantis dC18 色谱柱使用过程中的问题解决方案与故障排除HILIC Silica 色谱柱HILIC与反相色谱互补的选择性提高LC/MS灵敏度世界一流的宽PH值 极限色谱柱 Waters XTerra色谱柱 PH1-12 无与伦比的批间重现性 极佳的对称性 纯水流动相 Symmetry色谱柱100%纯水流动相 宽PH值 XTerra RP纯水极限色谱柱 PH2-12SunFire色谱柱 最好的低PH值稳定性 最佳粒度和批次重现性 满足制药行业最严格的性能要求
请问谁有4氨基安替比林分光光度法测定的酚类物质的标准曲线啊,是按照国标号HJ/T32-1999做的,固定污染源中酚类化合物的测定,我做了一个不知道哪里出了问题乱糟糟的,谁有的话给我一个看一下啊,找找区别,万分感谢啊
[b]手性(chirality)[/b]是三维物体的基本属性,三维结构的物体所具有的与其镜像的平面形状完全一致,但在三维空间中不能完全重叠的性质,正如人的左右手之间的关系。具有手性的化合物即称为[b]手性化合物[/b],手性化合物除了通常所说的含手性中心的化合物外,还包括含有手性轴、手性平面、手性螺旋等因素的化合物。一般来说,如果分子既无对称面也无对称中心,分子就具有手性。手性分子绝对构型的确定是一个极其重要且长期存在的问题。目前确定手性分子绝对构型的方法主要有四类:(1) 有机化学法;(2) 核磁共振法;(3) X射线衍射法;(4) 光谱法,如旋光光谱法、圆二色谱、振动圆二色谱等。[b]1. 有机化学法[/b]有机合成是最早的确定分子手性的方法,主要为化学相关法。即将目标分子反合成分析,从初始已知手性的化合物开始,通过手性控制的有机化学反应,将其转化为目标化合物的方法,然后从他们旋光符号或者相应的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、液相色谱推导出其绝对构型。很多富有挑战性的复杂手性化合物的合成如今已被有机化学家们所攻克,然而有机合成始终是一项繁琐而辛苦的选择。[color=#000000][b]2. 核磁共振法(NMR)[/b][/color][color=#000000]NMR 技术是获的化合物结构的首选方法,其耦合常数和NOE谱图是获取化合物相对构型的重要手段,适用于刚性结构非对映体的构型确认。但是对于光学(对映)异构体而言,一般情况下其NMR谱的信号是相同的,即应用NMR 谱无法直接将其区分,也不能确定其绝对构型。近年来发展了一些间接方法,借助NMR法,通过手性样品的衍生物来测定对映异构体的绝对构型。[/color][color=#000000]在应用NMR法测定手性化合物绝对构型的方法中,以Mosher 法最为常用。即通过将样品衍生化为非对映异构体或类似于非对映体,测定样品分子与手性试剂反应后产物的[sup]1[/sup]H-NMR 或[sup]13[/sup]C-NMR 位移数据,得到其化学位移的差值并与模型比较,最后推定底物分子手性中心的绝对构型。例如,Mosher法是将待测样品的手性仲醇基(或仲胺基)与([i]R[/i])或([i]S[/i])-α-甲氧基-α-三氟甲基-α-苯基乙酸(亦称Mosher 酸,缩写MTPA,见图1)反应生成相应的酯或酰胺,然后测定该酯或酰胺的核磁共振氢谱。根据MTPA芳香环的屏蔽效应,比较待测物与MTPA成酯(或酰胺)前后[sup]1[/sup]H-NMR 或[sup]13[/sup]C-NMR信号的化学位移差,由谱中化学位移的差值和模型图来推测仲醇(或仲胺基)的绝对构型。[/color][align=left]手性衍生物的NMR法的样品用量少,衍生物合成简单,测定迅速、准确,在手性醇、手性胺、手性羧酸的绝对构型确定中已经非常成熟。由于目前所开发的手性识别剂主要针对于手性中心中的某些基团(如羟基、氨基、羧酸),并且需要昂贵的手性试剂进行衍生化,其应用范围有所局限。 [/align][color=#000000][b]3. X-射线衍射法(X-raydiffraction)[/b][/color][color=#000000]普通的X-射线法(钼靶)仅能构筑化合物的相对构型,不能区分对应异构体。如果分子中含有重原子(一般原子序数大于16)或在分子中引入一个重原子,就可用X-射线来测定该重原子的手性分子绝对构型。此外,通过引入另一个已知绝对构型的手性分子也可获得结构的绝对构型。随着技术的发展,采用CuKa作为入射光源的X-射线单晶CCD衍射仪,对于测定相对分子量在1000以下、含C、H、N、O原子有机分子的绝对构型已可实现了。[/color][color=#000000]在单晶结构分析中,目前国际公认表征绝对构型的参数称为Flack 参数,当结构分析进入到最后的精修阶段时,如果该参数等于或接近0,或其参数在± 0.3之内,那么一般认为绝对构型就被确定了。[/color][color=#000000]采用单晶X-衍射法样品用量少、测定迅速、结果可靠直观,可以作为最终的立体构型的确定方法。但是由于测试的仪器价格昂贵,对单晶有严格要求,也限制了X-射线衍射法的应用。[/color][color=#000000][b]4. 光谱法[/b][/color][color=#000000]在光谱分析方法中,现有最有名和应用最广泛的手性分子构型确定法为旋光光谱法(ORD) 和圆二色谱法 (CD),该法对样品要求不高 (如纯度、官能团、结晶等)、测量过程无损失,因而得到了广泛应用。近几年,振动圆二色谱法 (VCD)取得了巨大的发展,逐渐成为一项鉴定手性分子绝对构型的重要工具。[/color][color=#000000][b]4.1 旋光光谱法(ORD)[/b][/color][color=#000000]早期的手性光学法是旋光谱法。当平面偏振光通过手性物质时, 能使其偏振面发生旋转,这种现象称之为旋光。 用仪器记录通过手性化合物溶液的平面偏振光的振动面偏转的角度,即为旋光度α,我们平常所测定的旋光即为波长在589.6 nm的Na灯的黄光下的比旋光度。旋光度随波长的变化而变化就可获得旋光光谱(ORD)。[/color][color=#000000]在同系物中,相同的化学反应使旋光值按相同的方向改变,而不改变其旋光的方向,因此通过比较相关化合物的旋光性,可得到手性化合物的构型信息。在采用该方法测定药物绝对构型时,应与绝对构型已知且与待测药物结构相同或相似化合物,在相同的实验条件下测定旋光光谱,以保证比较结果的可靠性。[/color][color=#000000]相比圆二色谱法(CD)而言,CD谱形尖锐、简单明了、易于分析,ORD现已被现代手性光学技术CD所取代。[/color][color=#000000][b]4.2 圆二色谱法 (CD)[/b][/color][color=#000000]传统的圆二色谱所用的平面偏振光的波长范围一般在紫外区(200~400 nm)。手性化合物(溶液)在左旋和右旋圆偏振光的吸收系数(ε)之差随入射偏振光波长的改变而改变, 得到的图谱即是圆二色光谱(CD),又称为电子圆二色谱(ECD)。[/color][color=#000000]该方法主要是通过测定光学活性物质(待测物)在圆偏振光下的Cotton效应,根据Cotton效应的符号获得药物结构中发色团周围环境的立体化学信息,并与一个绝对构型已知的与待测药物结构相似化合物的Cotton效应相比较,或者借助计算化学的方法,对比实验测值和理论计算值,即可能推导出待测物的绝对构型。[/color][color=#000000]长期以来,电子圆二色谱由于其干扰少、容易测定而被广泛应用。但该法使用的前提条件是待测化合物的手性中心含有合适的发色团(有紫外吸收),或者能够引进合适的发色团。对于手性中心无发色团或无法引入发色团的化合物,则不适宜采用该方法。[/color][color=#000000][b]4.3 振动圆二色谱法 (VCD)[/b][/color][color=#000000]传统的圆二色谱要求手性分子必须有紫外吸收,这一点成为限制其应用的重大问题。在20世纪70年代,Holzwart,Nafie和Stephens等先后成功测定了红外光区频率下的圆二色谱,即振动圆二色谱(VCD)。当平面偏振光的波长范围在红外区(4000~750 cm-1)时,由于其吸收光谱是分子的振动转动能级跃迁引起的,VCD谱即为红外光中的左旋圆偏光和右旋圆偏光的吸收系数之差∆ ε随波长变化所给出的图谱。[/color][color=#000000] 由于振动光谱谱图的复杂性, VCD很难象传统圆二色谱 (electronic circular dichroism, ECD)那样发展出合适的理论来进行结构-谱图的对应解释,主要依靠理论计算值和实测值对比来判断手性分子的绝对构型。[/color][color=#000000]与ECD相比,VCD的最大优势就是不需要分子中含有生色团 (紫外吸收),几乎所有手性分子都在红外区有吸收,都会产生VCD谱图。此外,VCD测试是在溶液状态测定,不需要单晶,样品中的非手性杂质也不影响测定结果。随着越来越多的关注和研究,振动圆二色谱法将成为一项鉴定手性分子绝对构型的强有力的工具。[/color][color=#000000]除上述的四大经典构型确定法外,红外光谱、紫外光谱法也用于辅助测定化合物的构型。更多的方法还望同行们共同探讨总结,希望大家在讨论区多多给予意见,谢谢![/color]本帖摘自“手性专家”微信公众号。
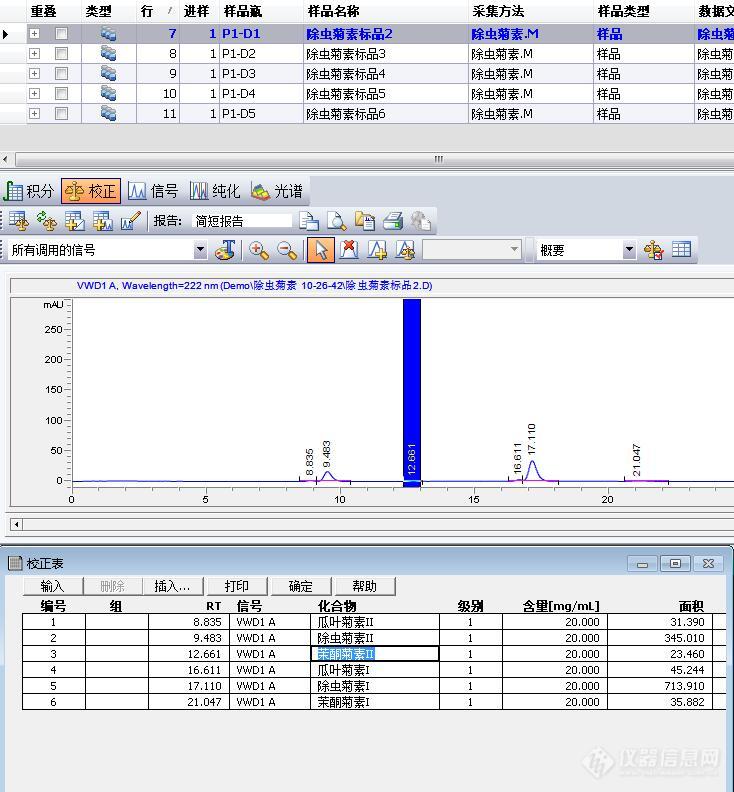
[align=center][size=18px]峰面积加和高阶用法之化合物组[/size][/align][align=left]在分析检测中,常常会有需要对多个化合物做汇总计算的情况。通常可以使用峰面积加和的方式计算。但对于多个化合物,化合物之间有其他峰的情况下,不能使用加和方式。此时可以使用化合物组来实现。下面就以除虫菊素为例,该产品包含6个化合物,分别是除虫菊素I,除虫菊素II,瓜叶菊素I,瓜叶菊素II,茉酮菊素I,茉酮菊素II,标品上只标注了6个化合物浓度总和,最终产品检测结果也只需要计算总浓度。[/align][align=left]1. 新建校正表,输入化合物浓度,这里输入总浓度20,并输入各个化合物名称[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301281424_6598_2963297_3.jpg[/img][/align][align=left]图1 建立校正表,输入浓度[/align][align=left][/align][align=left]2. 在校正表的“组”里输入组编号,本例中6个化合物编号都为1,然后在弹出的对话框里输入组名称“除虫菊素”,并勾选“组含量计算”,如图[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301280849_2653_2963297_3.jpg[/img][/align][align=left]图2 设置组编号及组名[/align][align=left][/align] 3. 继续添加新的级别,添加完成后,点击菜单栏校正/化合物组[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301285153_7838_2963297_3.jpg[/img]图3 打开化合物组4. 在化合物组细节中,在”组校正设置”中输入另外4个级别的浓度[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301283026_5851_2963297_3.jpg[/img]图4 输入各个级别的浓度5. 输入浓度后点确定,此时可以看到各个化合物按照面积百分比方式计算得到各自的浓度,并绘制标准曲线。[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301287402_7183_2963297_3.jpg[/img]图5 校正曲线(部分)6. 再设置报告格式为外标法,预览报告,即可得到样品各个化合物的浓度以及最后浓度加和[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301288534_1038_2963297_3.jpg[/img]图6 最终报告总结:此方法类似于MassHunter中的面积加和,只知道化合物总浓度,然后将面积加和后,根据峰面积百分比来计算各个化合物浓度,最后计算得总浓度。是面积加和的变种。
[color=#444444]最近合成了一个新化合物,是紫杉醇和一个小分子羧酸生成的酯,分子量1042。这个新化合物没人报道过怎样用液相测含量,紫杉醇含量测定大都是乙腈:水(1:1,V/V)。我的这个新化合物可以完全溶解在乙腈或者甲醇中。我用紫外扫过这个化合物最大吸收波长,发现它跟紫杉醇最大吸收波长几乎一样。请问我应该摸索这个化合物的色谱流动相条件,是用纯乙腈开始冲吗?非常感谢!![/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP