液相测雌三醇等7种性激素,一套有证标准物质下来多少钱?都有用哪些品牌?质控CRM怎么选择?
我用LC-MS 测定雌二醇、雌三醇、雌酮 ,在摸索质谱条件时 ,总找不到碎片离子 ,ESI+和ESI-都试过 参考文献用别人的质谱条件 ,做液质联用时不出峰 ,别人用的是C18的柱子 ,我们用的是C8 是柱子的问题吗 ?还是质谱的问题 ?我应该怎么解决呢
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
请教一下氰尿酸三钠盐的分析方法?
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测三氯乙醛废硫酸时,三氯乙酸乙酯和硫酸二乙酯峰重叠怎么办?或者色谱条件应该设为多少?谢谢!
查询检验20%的发烟硫酸游离三氧化硫的方法,都要用到玻璃安瓿球先进行溶液制备,这好像在一般的企业实验室很难做到,请问还可以用另外的简单快捷的方法测定吗?各位同行,你们用什么方法测酸度?
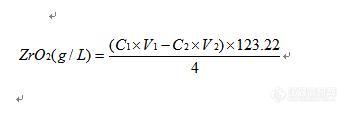
维权声明:本文为chenshaoj原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。现在硫酸锆溶液浓度分析方法大约有以下2种:1、 EDTA络合滴定法2、 重量法第一种方法是以二甲酚橙为指示剂,用EDTA标准溶液滴定至溶液由红色变为黄色即为终点,但通过长期运用,发现该方法指示剂指示终点颜色变化不明显,易照成较大误差;第二种方法是利用硫酸锆本身纯度很高,在配制的过程中又不用外加入任何化学物质即可溶解,所以取定量硫酸锆溶液,将其蒸发至干燥后,根据坩埚重量差即可计算出硫酸锆溶液的浓度,该方法较为准确,但大家都应该了解,重量法耗时很长,如果坩埚恒重不好的话,也容易造成较大误差;所以上述两种方法都具有一定的缺陷,不利于在生产中运用。所以我就改变思路,既然硫酸锆在配制的过程中并没有加入任何东西助溶,而硫酸锆又容易水解,其水解方程式如下:Zr(SO4)2+2H2O=2H2SO4+ZrO2 我们为何不能采用酸碱滴定其水解生成的硫酸从而计算出其氧化锆的浓度呢?所以就开始实验: 定量取一定体积的硫酸锆溶液(一般为2mL),加水稀释后以甲基红-亚甲基蓝为指示剂,用NaOH标准溶液滴定,溶液颜色由红色变为蓝色即为终点。 想法很好,但在实验的过程中发现了问题:由于在滴定的过程中,溶液的PH不断升高,所以硫酸锆也不断水解生成氧化锆,氧化锆会吸附指示剂,造成终点时变色不明显,而且颜色极易退掉,所以无法准确判断终点,换用其它酸碱指示剂(如酚酞等)也存在同样的问题。 既然溶液中的沉淀物会影响终点的判断,那为什么我们不把沉淀过滤后再来滴定呢?说干就干,通过实验发现,加入EDTA后硫酸锆也会水解(至今也没有想明白原理),而EDTA是中性化合物,不会对酸碱滴定产生误差,所以我就在硫酸锆溶液中加入EDTA溶液,等到硫酸锆全部水解后减压过滤,洗涤沉淀,滤液转移至三角瓶中以甲基红-亚甲基蓝为指示剂为指示剂,用NaOH标准溶液滴定,溶液颜色由红色变为蓝色即为终点;这次终点颜色很好判断,变色明显也不褪色,窃喜还以为成功了,但通过做几次平行样品后发现了问题:各次平行样品测定结果相差很大。这是什么原因呢?想来想去终于发现了问题:在加压过滤的过程中,溶液根本就没办法保证其全部将其水解生成的硫酸洗下来,而且EDTA是否会影响最后的滴定也不知道,另外这种方法也比较复杂,背离了我的初衷,所以该方法以失败告终。 后来又想了很多方法,比如直接用EDTA络合滴定时采用络黑T为指示剂等,最后发现也是存在指示终点不明显的老问题。 那我们可不可以采用反滴定法呢?那就用实验来说话吧:在硫酸锆溶液中定量加入过量的NaOH标准溶液,这时由于溶液呈碱性,硫酸锆早就全部水解了,而且溶液有一部分过量的NaOH,不过滤,以酚酞为指示剂,用盐酸标准溶液滴定至溶液红色刚好消失即为终点。通过实验,发现终点变色很明显,几个平行样品分析做出来结果基本一致,而且数据和定量配制的硫酸锆浓度刚好吻合。哈哈,实验成功,经过整理的方法如下:1、 原理硫酸锆水解后生成硫酸和氧化锆,先在硫酸锆溶液中定量的加入过量的氢氧化钠标准溶液,将水解出来的硫酸全部中和,过量的氢氧化钠以酚酞为指
国标上发烟硫酸中的三氧化硫含量是用酸碱滴定法测定的。我想请教下,有没有别的方法,是否有仪器可以进行测定。
我做有机酸,一直是用甲醇衍生化成酯方法,硫酸做催化剂,1~2小时回流。最近从《色谱分析样品处理》(也是从仪器网下的)看到,用三氟化硼作催化剂反应可在室温下很快完成。不知具体怎样?请各位做过的大虾具体指点一下,另外三氟化硼用的是纯的三氟化硼试剂还是三氟化硼络合物?
过硫酸钾的氧化能力应该足够把三价铬(三氯化铬)氧化成六价铬。今天做了个实验,出现了一个比较怪的结果:对于浓度分别为0,0.1,0.2,0.3,0.4mg/L的三价铬用过硫酸钾闭管氧化,然后用二苯碳酰二肼显色结果0.1,0.2mg/L的显色正常,而0.3,0.4mg/L的不显色,经检测,显色酸度正常。重复实验,出现同样结果。各位帮忙分析下原因
求 GB-T 11198.1-1989 工业硫酸 硫酸含量的测定和发烟硫酸中游离三氧化硫含量的计算 滴定法 非常感谢
谁知道用什么方法测定硫酸亚铁中三价铁吗?要考虑的干扰物质有铜、钴、锰等.还有预混料中三价铁的测定方法 ?谢谢
如题,双氯甲醚与2,4,6-三氯苯酚钠盐在一定条件下反应生成什么呢?(已知氯甲醚与2,4,6-三氯苯酚钠盐在一定条件下反应生成a-甲氧基三氯苯甲醚)
硫酸羟胺和三价铁反应是生成氧化二氮还是氮气?
同一瓶标液,用浓硫酸磺化和直接进样,响应值变低非常多,但是国标5009.176就是用浓硫酸,这是什么原因[img]https://ng1.17img.cn/bbsfiles/images/2019/04/201904231920250002_7710_3665228_3.png[/img]
对苯二胺硫酸盐、间苯二胺硫酸盐、邻苯二胺硫酸盐,用液相色谱C18柱能用吗?应配怎样的流动相? 它们的稀水溶液PH值在2-3左右。我用硫酸调流动相PH到2左右出两个峰。 又用三乙胺调PH到7多点还是两个
三氯化铁和硫酸铜在液相条件下能出峰吗? 谢谢色谱柱 C18流动相 乙腈: 缓冲液 (10%四丁基氢氧化铵溶液6ml,加水至200ml,磷酸调节PH为6.5):水(20:20:60)检测波长254NM,流速1请问在这种体系 下 硫酸铜和三氯化铁 会出峰吗:为什麽?
硫酸二甲酯是农药、染料、医药、香料工业等有机合成中广泛应用的甲基化剂。用以制造甲酯、甲醚、甲胺等。是二甲基亚砜、咖啡因、可待因、安乃近、氨基吡啉、甲氧苄氨嘧啶、香草醛以及农药乙酰甲胺磷等的原料。还可用作提取芳香烃类的溶剂。曾被用作战争毒剂。农药制造业硫酸二甲酯可用于有机磷杀虫剂、其他杀菌剂、其他除草剂等农药合成等。因为硫酸二甲酯作为一种重要的烷基化剂,在有机合成中常用于代替卤代烃作为甲基化试剂,进行O-甲基化反应和N-甲基化反应,可以用于诸如农药甲胺磷、乙酰甲胺磷、抗蚜威、氟蚜螨等杀虫杀螨剂的合成。但是硫酸二甲酯在高度高残留农药方面的应用市场处于相对萎缩的趋势,中国于2007年1月1日全面禁止甲胺磷等5种有机磷农药在国内农业上的使用,氟蚜螨等新型高效农药新品种,需要在技术上降低产品生产成本,而且需要做好登记产品上的推广应用工作。有机化工原料硫酸二甲酯可以用于醚类、醛类等有机化工原料的合成。例如重要的有机化工原料、溶剂和有机合成中间体——芳香醚,其最基本的制备方法是通过威廉姆逊合成法,其中硫酸二甲酯可作为甲基化试剂与苯酚反应合成芳香醚,主要反应历程分两步,首先苯酚与碱反应得到苯酚钠,生成的苯酚钠盐再与硫酸二甲酯反应合成苯甲醚。该反应为非均相反应。该过程的优点是硫酸二甲酯的价格相对其他甲基化试剂价格低廉,缺点是硫酸二甲酯的甲基化反应工业上采用低温间歇操作法生产,导致生产效率低、能耗高、劳动强度大,而且硫酸二甲酯有较强的毒性且致癌,对人的身体健康带来了隐患,再加上生产过程中产生大量工业废水,对环境污染严重。染料制造业硫酸二甲酯可用于阳离子染料、活性染料合成等。例如以硫酸二甲酯为甲基化试剂合成间甲基苯甲醚,间甲基苯甲醚主要用于以2-苯氨基-3-甲基-6-二丁氨基荧烷(ODB-2)为代表的荧烷类热敏染料的合成。催化剂及助剂硫酸二甲酯可以用于合成光稳定剂等助剂和催化剂。例如在50℃左右的环境下,往硬脂酸和三乙醇胺为原料合成的双长链酯胺有机溶液中,以一定速度滴加甲基化试剂硫酸二甲酯,可以合成酯基季铵盐,这是一种阳离子柔软剂,这种阳离子柔软剂有优良的柔软性能和较好的抗静电性,而且能够在环境中生物降解,比传统的双十八烷基二甲基氯化铵柔软剂环保。用硫酸二甲酯合成产物色泽乳白,处理织物后白度良好,织物柔软性能良好,只是硫酸二甲酯有剧毒,合成时候需要控制好用量,避免未反应的硫酸二甲酯残留在织物上对人造成损害。塑料制造业硫酸二甲酯在高分子领域可用于聚砜单体合成。也可用于塑料改性,例如硫酸二甲酯可以将三聚氰胺-甲醛树脂分子中的叔胺季铵化,从而在大分子链上引入离子,这样可以使高分子具有一定的导电性,从而制得结构型抗静电塑料。日用化学产品硫酸二甲酯在日化领域可用于照相乳剂制备、溶化,感光材料涂布,酚类、醚类、醛类香料合成,硝基麝香合成等。例如以氯仿为溶剂,缓慢往邻苯二酚中滴加碱性硫酸二甲酯,水浴加热一段时间,可以使邻苯二酚高效转化为愈创木酚,愈创木酚是香料香兰素的合成原料。医药工业硫酸二甲酯可用于合成药烃化、酰化、醚化等。 例如可以用于丙酮肟在碱性条件下甲基化为O-甲基丙酮肟,最后生成甲氧胺盐酸盐,这是一种重要的化工和药物中间体。可以用于杀菌剂苯氧菌酯的生成;在药物合成中,它可用于生产头孢地尼、头孢呋辛等药物。DNA甲基化硫酸二甲酯可以特异性使DNA中的G甲基化,实现DNA的化学修饰,而不影响其他的,也不影响RNA,DNA甲基化以后会导致基因表达被抑制,同时会导致被修饰的DNA在甲基化的位置断裂,实现DNA的化学裂解 ,传统的DNA测序方法之一:Maxam-Gilbert DNA化学降解法,就是利用DNA化学裂解后产生一系列片段,通过判断断裂位置的碱基或碱基类型,从而实现DNA的测序。
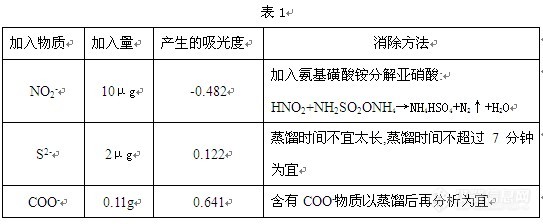
[color=#d40a00][size=2]维权声明:本文为[color=#ba4b01]abcdefghijkl123[/color][/size][color=#d40a00][size=2]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/size][/color]对于一些样品中亚硫酸盐含量的测定,你们是如何测定的,如何提高样品检测的准确性啊[/color] 食品中亚硫酸盐的检测方法国标为GB/T5009.34-2003。本文主要阐述了食品中亚硫酸盐的四种检测方法,并通过选取了有代表的样品将这四种检测方法与国标法进行了比较和探讨。从而证实了这四种检测方法对食品中亚硫酸盐的准确定量具有一定的实践指导意义。 亚硫酸及其盐类是广泛使用的食品添加剂,具有漂白、脱色、防腐及抗氧化等作用。国标GB/T5009.34-2003规定了食品中的亚硫酸盐检测方法:第一法为盐酸副玫瑰苯胺比色法及第二法蒸馏后碘量滴定法。但随着食品添加剂滥用现象严重,着色剂、食品中未知成分干扰等因素,采用GB/T5009.34-2003中规定的方法检测食品中亚硫酸盐仍存在着检测误差和很多的技术盲点,并特别选取了几种有代表性的食品来分析其亚硫酸盐含量,并做比较,认为有必要对其国标法进行改进和深入探讨。 一、检测方法: 第一法 盐酸副玫瑰苯胺法(蒸馏后比色法) 1.原理:亚硫酸盐与四氯汞钠反应生成稳定的络合物,再与甲醛和盐酸副玫瑰苯胺生成紫红色的络合物,比色定量。 2.试剂 2.1 盐酸副玫瑰苯胺:称取精制提纯(或进口分装)试剂盐酸副玫瑰苯胺10mg,加1+1盐酸15mL,用水溶解定容至50mL,该试剂保存于冰箱,1周内稳定。 2.2 氨基磺酸铵: 12g/L 2.3 甲醛: 2g/L 2.4 SO2标准贮备液配制: 先测定固体试剂NaHSO3中SO2的纯度,而后根据已知SO2的纯度,直接称取NaHSO3配成SO2标准溶液,使用时无需再进行标定。这样标定之后的固体NaHSO3试剂可使用1年,用0.1moL/LNaOH溶液配成1000μg/mL贮备液,该贮备液于冰箱内至少稳定1个月。 2.5 SO2标准使用液配制:临用前用四氯汞钠吸收液稀释成2μg/mL的标准使用液。 3.方法 试样处理:在密闭容器中加1+1盐酸对试样进行酸化并加热蒸馏,释放出的SO2以四氯汞钠作为吸收液,定容至一体积,吸取一定量的试液分析。后面步骤及计算同GB/T5009.34-2003第一法。 4.方法探讨 4.1 干扰物质及消除方法见表1[img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008091755_235403_2961690_3.jpg[/img] 4.2 COO- 干扰测定,会与盐酸副玫瑰苯胺形成紫红色络合物,典型样品如味精(谷氨酸钠分子结构中含有COO-),须采用蒸馏后比色,可排除此干扰,否则数据严重虚高。 4.3含硫基食品(如大蒜、萝卜、姜、辣椒、木耳等)蒸馏时间不超过7分钟为宜,蒸馏时间过长,则硫基化合物开始馏出,必要时对蒸馏装置进行改进,否则会干扰测定,数据虚高。 4.4 比色法适用于SO2含量低于0.1g/kg试样的检测。 第二法 蒸馏后碱滴定法 1原理 亚硫酸盐在酸性条件下用碱中和,加热,亚硫酸盐被过氧化氢吸收,用碱中和并滴定至终点,根据消耗标液用量计算其含量。 H2O2+H2SO3→H2SO4+H2O H2SO4+2NaOH→Na2SO4+2H2O 2试剂: 2.1 盐酸1+1 2.2 过氧化氢1+10 2.3 氢氧化钠标准溶液 0.05moL/L 2.4 甲基红-亚甲基蓝指示剂 3步骤 蒸馏过程同第一法,另在接收器内加入过氧化氢(1+10)20mL,加指示剂1滴,用0.05moL/L NaOH标液中和至灰绿色终点,作为吸收液,蒸馏至一定体积后,取下吸收液,再加一滴指示剂,用0.05moL/L NaOH标液滴定至灰绿色为终点。 4计算公式 X(g/kg)=C*V*32/m X:样品中二氧化硫的含量, g/kg C: 0.05moL/L NaOH标液浓度 V:滴定过氧化氢吸收液至终点时NaOH标液消耗毫升数 32:与1mLNaOH标液相当的二氧化硫的毫克数 5方法讨论 5.1该法与国标第二法相比,方法灵敏度更高,选择性相对更强,所须药品更简单,易于操作。 5.1该法适用于二氧化硫含量在0.1 g/kg以上试样的检测。 第三法 盐酸副玫瑰苯胺比色法(加过氧化氢) 适用范围:当检测带色的发酵酒、蒸馏后馏出液含有颜色、红糖等样品时,按GB/T5009.34-2003第一法,样品比色管均呈现一定量底色而干扰测定,使得SO2测得数据虚高,采用加入H2O2法消除呈色物质的背景干扰可得较为满意结果。 1. 原理:H2O2将SO2氧化成硫酸后,不与显色剂盐酸副玫瑰苯胺显色,且在波长550nm下不产生吸光度,呈色物质所产生的吸光度作为底液,予以扣除。 2.试剂 2.1 0.3% H2O2 2.2 其它试剂同本文第一法 3. 步骤:样品不须蒸馏,同GB/T5009.34-2003第一法处理。 显色:同GB/T5009.34-2003第一法,只是在测定样品时,须同时吸取2份样液于A、B管中,A管加0.2mL水,B管则加入0.2mL0.3% H2O2,它们所产生的吸光度相减所得的吸光度(A-B)作为样液中SO2所产生的吸光度,代入计算。 第四法 [url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法 1.原理:试样中的亚硫酸在20%磷酸酸性条件下,于90℃水浴中通氮曝气分离,收集在三乙醇胺溶液中,用IC测定。 2.试剂 2.1 亚硫酸标准溶液:按本文第一法配制成100μg/mLSO32-标准原液。用时用吸收液将原液稀释成0.1-4.0μg/mLSO32-使用液 2.2 吸收液:10克三乙醇胺加水溶解至1L 2.3 1%氨基磺酸铵 2.4 洗脱液:3.71克碳酸钠和3.36克碳酸氢钠加水溶解至1L,用时用水稀释10倍,碳酸钠和碳酸氢钠的浓度分别为3.5mmoL/L和4.0mmoL/L 2.5 净化剂163.3克十二烷基苯磺酸加水溶解至1L,用时用水稀释10倍 2.6 2,4-二硝基苯肼(2,4-DNP)液:1克2,4-DNP加40%磷酸溶解至100mL 3.仪器及测定条件: 3.1 ICS-1500[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]:美国戴安公司 3.2 亚硫酸收集装置 3.3 色谱柱:IonPac AS14分析柱, 250mm×4mm 3.4 洗脱剂:3.5mmoL/LNa2CO3/4.0mmoL/LNaHCO3 洗脱剂流速:2.0mL/min 3.5 净化剂:50mmoL/L十二烷基苯磺酸,净化剂流速:2.0mL/min 3.6 检测器:抑制型电导检测器 3.7 温度:40℃ 3.8 进样量:100μL 4.操作方法 4.1 试验溶液的制备:取10mL 吸收液加入A吸收管内,5mL2,4-DNP液加入联接SO2收集装置的B管内。取1~5克试样放入已通氮脱气10分钟的40mL20%磷酸和1mL氨基磺酸铵溶液的曝气瓶内,立即接好收集装置。曝气瓶于90℃水浴中以800mL/min流速通氮曝气40min,所得吸收液即为试验溶液。用水代替试样同上操作,得到的吸收液为空白试验溶液。 4.2 测定:取100μL试验溶液和空白试验溶液注入IC内测定亚硫酸离子的峰面积,根据预先用标准溶液(0.1-4.0μg/mLSO32-)作成的标准曲线计算试样中二氧化硫含量。 5 说明:本法标准曲线,亚硫酸在0.1-4.0μg/mL范围内呈线性关系,检出限为0.2mg/kg。1000mgCl-、SO42-、NO3-、甲酸、乙酸、丙酸、乙醇、10mg甲醛、糠醛、二甲基硫醚、二甲基二硫醚、甲硫醇钠、丙烯基异硫氰酸盐及0.1mgS2-对1μg/mL亚硫酸无干扰。NO2-、乙醛、苯甲醛有严重负干扰,但NO2-干扰用氨基磺酸铵,醛类干扰用2,4-DNP消除。 二、总结: 1.本文介绍的四种检测方法与国标法比较结果见表2。由表可见,对于一些颜色较浅的食品如白木耳、粉丝、白糖、竹荪等,这些检测方法与国标法相一致,但一些颜色较深的样品和含有羧基的样品,干扰因素较大,用国标法检测本来不含有SO2的样品,却也测出了较高的值。而用本文介绍的检测方法可得较为满意的数值。[img]http://ng1.17img.cn/bbsfiles/images/2010/08/201008091756_235404_2961690_3.jpg[/img] 2. 样品制备时,尤其是干类果实的个体差异较大,故要注意样品的均质化。 3. 这四种的检测方法,可根据样品的实际情况,结合采用其中的检测方法进行食品中亚硫酸盐含量的测定。
我根据国标在做二氯乙酸、三氯乙酸的检测项目,但是衍生出来的东西好像是有问题的,请各位看看我的衍生条件有没有什么问题:取25ml水,加入1.5ml浓硫酸,一勺硫酸钠,内标和标准,之后加入4ml左右的甲基叔丁基醚,震摇。取上清液3ml,再加入现配的衍生试剂(浓硫酸:甲醇为1:9)2.5ml。之后放在50℃水浴锅中恒温水浴2h,取出来4℃冷却。加入4ml碳酸氢钠,剧烈震摇,取上清液,上气相检测。请各位帮我看看有什么问题,做的时候需要什么注意事项!!!多谢
[font=SimSun, STSong, &]我国GB 2760规定“凡列入合成香料目录的香料,若存在相应的铵盐、钠盐、钾盐、钙盐和盐酸盐、碳酸盐、硫酸盐,且具有香料特性的化合物,应视作已被批准使用的香料。”[/font][font=SimSun, STSong, &]国外以及codex等国家也有类似规定吗?比如codex把肉桂酸视作香精,那肉桂酸钾是否也是香精香料呢?有规定出处吗?[/font]
我按药典的配制方法:取碱式品红0.2g,加热水100ml溶解后,放冷,加亚硫酸钠溶液 (1→10)20ml、盐酸2ml,用水稀释至200ml,加活性炭0.1g,搅拌并迅速滤过,放置1小时以上,即得。 本液应临用新制。我配了好多次配出的都是黄色的。请教各路神仙,问题出在哪里?我用焦亚硫酸钠和无水硫酸钠都试过了,都不行。在加入亚硫酸钠溶液后一会儿就出现很多沉淀,加入盐酸后沉淀都消失了。此时产生的沉淀是什么?我配这个试剂是为了检测白蜂蜡的丙三醇与其他多元醇项目丙三醇和其它多元醇 取本品0.20g加氢氧化钾乙醇溶液(取氢氧化钾3g,加水5ml使溶解,加乙醇至100ml,摇匀,即得)10ml,加热回流30分钟,取出,加稀硫酸50ml,放冷,滤过,用稀硫酸洗涤容器和滤器,合并洗液和滤液,置同一100ml量瓶中,加稀硫酸稀释至刻度,摇匀,作为供试品溶液。取10ml纳氏比色管两支,甲管中精密加入供试品溶液1ml,加0.05mol/L高碘酸钠溶液0.5ml,混匀,放置5分钟,再加品红亚硫酸试液1.0ml,混匀,不应出现沉淀;然后将试管置40℃温水中。在水温下降过程中不断旋转试管,观察10~15分钟;乙管中精密加入0.001%丙三醇的稀硫酸溶液1ml,与甲管同时依法操作,甲管中显出的颜色与乙管比较,不得更深。(以丙三醇计,不得过0.5%)加入我配的品红亚硫酸钠样品盒对照就会出现沉淀。不知产生的是什么沉淀?问题出在何处?当我加两毫升品红亚硫酸溶液时沉淀消失,这又是为什么?我已经被这个实验困了好久了,请教大家,帮帮忙啊,感激不尽!
发烟硫酸中SO3的测定方法本方法为GB11198.1-89《工业硫酸 硫酸含量的测定和发烟硫酸中游离三氧化硫含量的计算 滴定法》。此标准参照采用国际标准ISO910-1977《工业硫酸和发烟硫酸——总酸度的测定和发烟硫酸中游离三氧化硫含量计算——滴定法》。 1.1 方法原理 以甲基红-次甲基蓝为指示剂,用氢氧化钠标准溶液中和滴定,以测得硫酸含量。或由测得的硫酸含量换算成游离三氧化硫含量。 1.2 试剂和溶液 氢氧化钠(GB629)标准溶液:c(NaOH)=0.5mol/L;甲基红-次甲基蓝混合指示剂。 1.3 仪器 玻璃安瓿球(直径约15mm,毛细管端长约60mm)。 1.4 称样和试液的制备 1.4.1 特种硫酸和浓硫酸 用已称量的带磨口盖的小称量瓶,称取约0.7g试样(称准至0.0001g)小心移入盛有50ml水的250ml锥形瓶中,冷却至室温,备用。 1.4.2 发烟硫酸 将安瓿球称量(称准至0.0001g),然后在微火上烤热球部,迅速将该球之毛细管插入试样中,吸入约0.7g试样,立即用火焰将毛细管顶端烧结封闭,并用小火将毛细管外壁所沾上的酸液烤干,重新称量。 将已称量的安瓿球放入盛有100ml水的具磨口塞的50ml锥形瓶中,塞紧瓶塞,用力振摇以粉碎安瓿球,继续振荡直至雾状三氧化硫气体消失,打开瓶塞,用玻璃棒轻轻压碎安瓿球的毛细管,用水冲洗瓶塞、瓶颈及玻璃棒,备用。 1.5 测定步骤 1.5.1 特种硫酸的浓硫酸 于试液(1.4.1)中,加2-3滴混合指示剂,用氢氧化钠标准溶液滴定至溶液呈灰绿色为终点。 1.5.2 发烟硫酸 于试液(1.4.2)中,加2-3滴混合指示剂,用氢氧化钠标准溶液滴定至溶液呈灰绿色为终点。 1.6 计算 1.6.1 特种硫酸和浓硫酸 硫酸的含量X(%)按式(1)计算: X=(V*c*0.04904)/m*100 (1)式中 V——滴定耗用的氢氧化钠标准溶液体积,ml; c——氢氧化钠标准溶液浓度,mol/L; 0.04904——与1.00ml1.000mol/L氢氧化钠标准溶液相当的,以克表示的硫酸的质量。 1.6.2 发烟硫酸 发烟硫酸中游离三氧化硫的含量X1(%)按式(2)计算或由附录A表A1查得。 X1=4.444*(X-100) (2)式中 X——按1.6.1条中式(1)算出的发烟硫酸中硫酸的质量百分含量; 4.444——游离三氧化硫的换算系数。 1.7 允许误差 测定结果以算术平均值报出。 1.7.1 特种硫酸和浓硫酸中硫酸含量平行测定允许绝对偏差为0.2%。 1.7.2 发烟硫酸中游离三氧化硫含量平行测定允许绝对偏差为0.6%。
2010版药典白蜂蜡新增了丙三醇和其它多元醇项目 取本品0.20g加氢氧化钾乙醇溶液(取氢氧化钾3g,加水5ml使溶解,加乙醇至100ml,摇匀,即得)10ml,加热回流30分钟,取出,加稀硫酸50ml,放冷,滤过,用稀硫酸洗涤容器和滤器,合并洗液和滤液,置同一100ml量瓶中,加稀硫酸稀释至刻度,摇匀,作为供试品溶液。取10ml纳氏比色管两支,甲管中精密加入供试品溶液1ml,加0.05mol/L高碘酸钠溶液0.5ml,混匀,放置5分钟,再加品红亚硫酸试液1.0ml,混匀,不应出现沉淀;然后将试管置40℃温水中。在水温下降过程中不断旋转试管,观察10~15分钟;乙管中精密加入0.001%丙三醇的稀硫酸溶液1ml,与甲管同时依法操作,甲管中显出的颜色与乙管比较,不得更深。(以丙三醇计,不得过0.5%) 大家有没有做成功的?我在检测时对照和样品都出现沉淀,不知是怎么回事?
请问,酸铜中的氯离子 光铬中的硫酸根离子三价铬离子各用什么仪器测? 价格怎样?哪一家公司的好用?怎样联系?谢谢各位了!
2010版药典白蜂蜡新增了丙三醇和其它多元醇项目取本品0.20g加氢氧化钾乙醇溶液(取氢氧化钾3g,加水5ml使溶解,加乙醇至100ml,摇匀,即得)10ml,加热回流30分钟,取出,加稀硫酸50ml,放冷,滤过,用稀硫酸洗涤容器和滤器,合并洗液和滤液,置同一100ml量瓶中,加稀硫酸稀释至刻度,摇匀,作为供试品溶液。取10ml纳氏比色管两支,甲管中精密加入供试品溶液1ml,加0.05mol/L高碘酸钠溶液0.5ml,混匀,放置5分钟,再加品红亚硫酸试液1.0ml,混匀,不应出现沉淀;然后将试管置40℃温水中。在水温下降过程中不断旋转试管,观察10~15分钟;乙管中精密加入0.001%丙三醇的稀硫酸溶液1ml,与甲管同时依法操作,甲管中显出的颜色与乙管比较,不得更深。(以丙三醇计,不得过0.5%)大家有没有做成功的?我在检测时对照和样品都出现沉淀,不知是怎么回事?
硫酸草酸酒石酸三酸体系怎么测各种的浓度?大概是60g/L硫酸,40g/L酒石酸,20g/L草酸
火焰法原子吸收测硫酸钠中的铅,钠盐会产生干扰吗?
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。
硫酸草酸酒石酸三酸体系怎么测各种的浓度?大概是60g/L硫酸,40g/L酒石酸,20g/L草酸







 我要推广仪器
我要推广仪器
 下载APP
下载APP