N,N'-二琥珀酰亚胺基碳酸酯能用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测吗?
请问乙二胺二琥珀酸(EDDS)怎么溶解,有没有人知道,感觉在水中不溶
我需要药品[S,S]-EDDS,中文名是乙二胺二琥珀酸,请与我联系bluebell22@tom.com
请问专家:我急需要做黑索今(三次甲基三硝胺,一种六元环形化合物的高能炸药,环上带3个硝基)的粒度分布,用的分散剂是二辛基琥珀酸磺酸钠,分散介质用的是水,平行结果不好,请问有没有什么合适的分散剂和分散介质?-我用的是美国库尔特公司的LS-230激光粒度仪。——盼回复,不胜感激!
ICP-AES一般是检测无机元素含量,主要是金属元素。我现有个疑问,亚胺基二乙酸想要检测一下其中的元素含量,并且它可以溶于水,那么水溶解以后是否就可以上机检测了?因曾做过这一类型的有机物,进样到ICP-AES中会熄火,不知道是不是因为有机物的原因,所以不太敢检测亚胺基二乙酸,检测有机物对仪器影响大吗?会不会把仪器烧了?请各位支招。谢谢!
【序号】:1【作者】: 张雅伦【题名】:聚乙二醇的N-羟基琥珀酰亚胺琥珀酸酯的合成工艺、苏氨酸负载及催化应用【期刊】:兰州大学【年、卷、期、起止页码】:2017【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C475KOm_zrgu4lQARvep2SAkOTSE1G1uB0_um8HHdEYmZhkBIZJEK02VaOdneXeYijuWwpOfpIhlJTd0mjIpAyz7&uniplatform=NZKPT

2010年版药典(一部)中,对益母草中盐酸水苏碱的测定有如下描述(以丙基酰胺键合硅胶为填充剂):http://ng1.17img.cn/bbsfiles/images/2011/01/201101080907_272670_801_3.jpg那么为什么要用丙基酰胺柱来测盐酸水苏碱呢?丙基酰胺硅胶基质的柱子是什么柱子呢? 首先我们要了解盐酸水苏碱的特性,盐酸水苏碱的极性极大,普通的反相色谱对它的保留分离能力较弱,通常在死时间里流出而无法得到分离,而亲水作用色谱HILIC能为极强性的化合物提供良好的保留,在此类化合物上应用广泛。 目前已有多种商品化的HILIC色谱柱,多为硅胶基质,键合不同极性基团,如丙基酰胺基,酰胺基,聚琥珀亚酰胺等极性基团,氨基键合硅胶柱由于使用寿命较短,键合相容易流失,造成保留 丙基酰胺键合硅胶克服了传统正相色谱柱在水相条件下不稳定的缺点,其常使用流动相是和反相色谱相同的水相缓冲液( 40%)及有机溶剂,但是其梯度条件通常是初始为高比例有机相,逐步加大水相含量;极性丙基酰胺键合硅胶的HILIC色谱柱在反相条件下,可以有效的保留极性化合物,是一种崭新的极性化合物HPLC分离解决方式.博纳艾杰尔推出的Venusil HILIC (丙基酰胺键合硅胶),就是一样一款非常适合于益母草中盐酸水苏碱测定的柱子,测定方法及谱图如下:色谱柱:Venusil HILIC (丙基酰胺键合硅胶),4.6×250mm,5µm,100Å(订货号:VH952505-0)流动相:乙腈-0.2%冰醋酸(80:20)流速:0.5mL/min柱温:25℃进样体积:20μL检测器:ELSD蒸发光散射检测器http://ng1.17img.cn/bbsfiles/images/2010/11/201011291710_262707_801_3.jpg益母草供试品含量测定色谱图(主峰保留时间:22.697min)
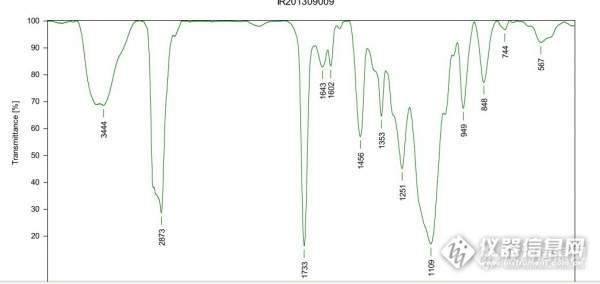
我的实验是这样的,我们在模仿国外一个丙烯酸酯聚合物的产品,从热裂解-GCMS的结果来看,里面还有丙烯酸丁酯,乙烯基吡啶等主要单体,但经过化学测试,里面胺基含量比较高,但图谱结果中却怎么也找不到含有胺基的化学物,所以一直很头疼,我想知道通过什么其他办法可以尝试知道里面的胺基到底是什么物质?谢谢!附件是该物质的红外!!http://ng1.17img.cn/bbsfiles/images/2013/11/201311181049_477794_2113729_3.jpg
二甲胺基磺酰氯在合成过程中在主峰前出现一个1%的左右的杂质,请教高手帮我解疑是什么?
最近看了一些文献,发现做氨基酸的方法非常多,这里罗列几种,不知道各位主要用哪种?由于大多数的氨基酸在紫外区没有吸收或只有微弱的吸收,为提高检测灵敏度和分离度,需要对氨基酸进行衍生化,这样才能被紫外或荧光检测器所检测。另外氨基酸极性非常大,无法用反相分析,通过衍生化也可以变离子型化合物为非离子型,用反相方法分离。1、 HPLC 柱前衍生柱前衍生:邻苯二甲醛(OPA)衍生一级氨基酸、9-氯甲酸芴甲酯(FMOC)衍生二级氨基酸色谱柱:AAA专用柱检测器:DAD(338nm OPA-氨基酸;262nm FMOC-氨基酸)2、 HPLC柱前衍生柱前衍生:异硫氰酸苯酯(PITC)色谱柱:AAA专用柱检测器:UV 254nm3、 HPLC柱前衍生柱前衍生:2,4-二硝基氟苯(DNFB)色谱柱:AAA专用柱检测器:UV 360nm4、 HPLC柱前衍生柱前衍生: 6氨基喹啉基N羟基琥珀酰亚氨基甲酸酯(AQC)色谱柱:反相柱(C18)检测器:UV 248 nm5、 HPLC 柱后衍生柱后衍生:邻苯二甲醛(OPA) 色谱柱:AAA专用柱检测器:荧光或紫外6、 离子色谱法:HPAEC-IPAD(高效阴离子交换分离-积分脉冲安培检测)7、氨基酸分析仪
由二甲胺与磺酰氯合成二甲胺基磺酰氯,产品计划用气相做检测,柱子是se-30的,结果不理想,大家给些意见,尤其是关于该产品的文献极少,在知网上没有几篇。也可能太简单了。
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
查了很多资料,好像没有使用液相单独测定琥珀酸的含量,一般都是测定有机酸的(同时测定几种有机酸,其中包括琥珀酸)。今天我按照参考文献做了琥珀酸的测定。条件如下色谱柱:Shimadzu C18柱(5um粒径 250×4.6mm)流动相:5% CH3OH – 0.10 mol/ L KH2PO4(pH 3.0)缓冲溶液(V/ V)流速:1 mL/ min 柱箱温度:室温(22 ±2 ℃) 检测波长:215 nm进样量:20μL资料说此条件下琥珀酸的保留时间为7-8min,线性范围0-600ug/mL.今天分别进了1mg/L,10mg/L的标样,可是20min内一直不出峰,怀疑是标样的浓度太低,后来又进了1mg/mL 的标样,结果20mi内仍然不出峰。(我是新手,当时没有继续延长时间.....)我以为此方法测不出来,后来用甲醇冲洗柱子时在60min左右出现了几个很大的峰。 我不知道是我的标样琥珀酸的峰,还是其他的物质?? 比如柱子里的其他残留物? 不过我觉得进样前柱子已经冲洗干净了哪位大侠测过琥珀酸的,能不能给些指导? 液相是不是不适合低或是微含量琥珀的测定。先跪谢 @ 哭谢!!
有报道一种HPLC测定生物样品中含硫化合物、氨基酸和生物胺的方法,该方法是采用磺酰氯对上述目标物进行柱前衍生,而后通过[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]柱分离,在460nm下进行监测。该方法的新颖之处是采用了磺酰氯进行衍生,与OPA相比,磺酰氯不仅能衍生伯胺,还能与仲胺反应,由于其活性较高,还能与羟基、苯酚、巯基等反应,用途较广。[font=-apple-system, BlinkMacSystemFont, &][color=#333333][back=#fcfcfc]该方法允许同时分析脑标本、尿液、血浆和细胞裂解物中的生物胺、氨基酸和磺氨基化合物,包括肌肽、多巴胺、肾上腺素、谷胱甘肽、半胱氨酸、牛磺酸、兰硫氨酸和胱硫硫氨酸。此外,该方法适用于研究牛磺酸和谷胱甘肽的生理和非生理衍生物,例如次牛磺酸,高牛磺酸,同型半胱氨酸和S-乙酰谷胱甘肽。详见DOI [url=https://doi.org/10.1007/978-94-024-1079-2_42]10.1007/978-94-024-1079-2_42[/url] [/back][/color][/font]
如何分析多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)中水分的含量(应该是几十个PPM)??如果用KF滴定法怎么消除其碱性太强的现象??如果用水杨酸酸中和的话是不是生成水???我们现用的分析方法是加水杨酸然后滴空白再加样品测定水含量.这种方法有问题吗???
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
二甲胺基磺酰氯在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]中出峰吗?
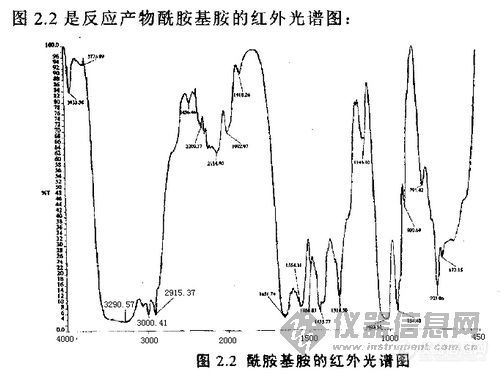
间苯二甲胺 和 己二酸反应生成 酰胺基胺,可是红外光谱上只能看见仲酰胺基的特征吸收峰,为什么看不到伯胺的峰?3000.41和2915.37处是不是伯胺吸收峰?是不是往后挪了?[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906080958_154498_1608859_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906080958_154497_1608859_3.jpg[/img]
[font=黑体]项目概况[/font][font=仿宋]非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒等试剂耗材采购招标项目的潜在投标人应在安徽公共资源交易集团电子交易系统或安徽合肥公共资源交易电子服务系统获取招标文件,并于2023年05月22日14点00分前递交(上传)投标文件。[/font][font=黑体]一、项目基本情况[/font][font=仿宋]项目编号:2023BFFHZ00759[/font][font=仿宋]项目名称:非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒等试剂耗材采购[/font][font=仿宋]预算金额:675.7304万元[/font][font=仿宋]最高限价:675.7304万元[/font][font=仿宋]采购需求:非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒等试剂耗材采购,详见招标文件[/font][font=仿宋]合同履行期限:合同签订后分批供货,每批货物接到采购人书面通知后5个工作日完成供货。[/font][font=仿宋]本项目是否接受联合体投标:否[/font][font=黑体]二、申请人的资格要求[/font][font=仿宋]1.满足《中华人民共和国政府采购法》第二十二条规定; 2.落实政府采购政策需满足的资格要求:无 3.本项目的特定资格要求:具有医疗器械生产(或经营)资格(含体外诊断试剂)。[/font][font=黑体]三、获取招标文件[/font][font=仿宋]1.时间:2023年04月29日09:00至2023年05月22日14:00[/font][font=仿宋]2.地点:安徽公共资源交易集团电子交易系统或安徽合肥公共资源交易电子服务系统[/font][font=仿宋]3.方式:(1)投标人须登录安徽公共资源交易集团电子交易系统或安徽合肥公共资源交易电子服务系统(以下简称“电子服务系统”)查阅招标文件。首次登录须持有电子服务系统兼容的数字证书,详情参见电子服务系统办事指南。 (2)招标文件获取过程中有任何疑问,请在工作时间(9:00-17:30,节假日休息)拨打技术支持热线(非项目咨询):4009980000。项目咨询请拨打电话:0551-66223922。[/font][font=仿宋]4.售价:免费[/font][font=黑体]四、提交投标文件截止时间、开标时间和地点[/font][font=仿宋]1.提交(上传)投标文件截止时间(开标时间):2023年05月22日14点00分[/font][font=仿宋]2.提交(上传)投标文件地点(开标地点):合肥市滨湖新区南京路2588号要素交易市场A区(徽州大道与南京路交口)2楼8号开标室[/font][font=黑体]五、公告期限[/font][font=仿宋]自本公告发布之日起5个工作日。[/font][font=黑体]六、其他补充事宜[/font][font=仿宋]1.本项目落实节能环保、中小微型企业扶持等相关政府采购政策。 2.本次公告同时在安徽合肥公共资源交易中心网站、安徽省政府采购网、安徽省公共资源交易监管网、全国公共资源交易平台上发布。 3.投标人应合理安排采购文件获取时间,特别是网络速度慢的地区防止在系统关闭前网络拥堵无法操作。如果因计算机及网络故障造成无法完成采购文件获取,责任自负。 4.本项目实施全流程电子化交易,投标文件实施网上远程解密,投标人无需前往开标现场。 5.本项目符合财政部、工业和信息化部制定的《政府采购促进中小企业发展管理办法》第六条第二款之规定,为非专门面向中小企业采购项目。具体原因如下:因确需使用不可替代的专利、专有技术,基础设施限制,或者提供特定公共服务等原因,只能从中小企业之外的供应商处采购的。如对此项内容有疑问,可按采购文件约定提出询问或质疑。[/font][font=黑体]七、对本次招标提出询问,请按以下方式联系[/font][font=仿宋]1.采购人信息[/font][font=仿宋]名称:合肥市妇女儿童保健中心[/font][font=仿宋]地址:合肥市庐阳区益民街15号[/font][font=仿宋]联系方式:18155168913[/font][font=仿宋]2.采购代理机构信息[/font][font=仿宋]名称:安徽公共资源交易集团项目管理有限公司[/font][font=仿宋]地址:合肥市滨湖新区南京路2588号(徽州大道与南京路交口)六楼[/font][font=仿宋]联系方法:0551-66223922[/font][font=仿宋]3.项目联系方式[/font][font=仿宋]项目联系人:李工[/font][font=仿宋]电话:0551-66223923[/font]
最近在做氨基酸分析,买了waters的氨基酸分析标准试剂盒(UPLC配套的),看有论坛版友说waters的分析方法包用的衍生剂是AQC(6-氨基哇琳基-N-琥珀酰-亚胺基甲酸酯)但是衍生方法感觉又和AQC的相关文献不同,不是到时什么情况?有比较了解的朋友麻烦帮忙解释一下,谢谢!
请问老师:如何分析盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量??含量在几个到几十个PPM
用TLC检测苯胺基乙腈和苯胺基乙酸钾,应该使用那种染色剂呢?基本的显色剂碘或者硫酸是否可以?
6-氨基喹啉基-N-羟基琥珀酰亚胺基甲酸酯(AQC)为衍生试剂,准备利用AQC做氨基酸分析?有做过的吗?是不是仅Waters有?其他公司有类似替代品吗?补充一下:大概300个样品。紫外检测
哪位大侠知道酰胺基丙酸的分析方法,先谢谢!在线等!
请问老师,如果用光谱的方法进行盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量的测定,它们的吸收峰是多少???
[color=#444444]苯胺基乙酸钾,原来一直使用重氮滴定,但是发现副产物影响,不能准确指导生产,如果用液相色谱分析,有没有朋友做过类似的分析,目前的基本思路是,加入缓冲溶液控制PH进C18柱,由于没有用缓冲盐的经验,不指导该用什么做缓冲溶液,流动相。最好能直接提供相关产品色谱分析文献。[/color]

【中文名称】琥珀酸柠檬酸铁钠;丁二酸亚铁合枸橼酸钠【英文名称】ferrous succinate sodium citrate;sodium ferricsuccinate citrate【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/04/201204192020_362339_1855403_3.jpg【毒性LD50(mg/kg)】 大鼠经口2200【性状】 青白至微带绿色的白色粉末,无臭,有微弱的铁味。有绿色的荧光。【溶解情况】 易溶于热水,溶液呈中性并显黄绿色,不溶于乙醇及其他有机溶剂。【用途】 用作食品的铁强化剂或饲料添加剂,用于调制奶粉、离乳食品以及缺铁病人、孕妇和产妇等食品的强化。【制备或来源】 用柠檬酸、琥珀酸、硫酸亚铁、碳酸钠溶液为原料制得。【其他】 略
[color=#444444]本人最近按2015版药典做了一个药用辅料-醋酸羟丙甲纤维素琥珀酸酯的的游离乙酸的含量测定实验。实验过程如下:[/color][color=#444444] 游离乙酸、琥珀酸 取本品0.102g,精密称定,置锥形瓶中,精密加入磷酸盐溶液(取0.02mol/L磷酸二氢钾溶液,用1mol/L氢氧化钠溶液调pH值至7.5)4.0ml,搅拌2小时,加磷酸溶液(取1.25mol/L磷酸1ml,置50ml量瓶中,加水稀释至刻度,摇匀)4.0ml,强力振摇,离心,上清液作为供试品溶液;精密称取琥珀酸0.13g,置100ml量瓶中,加水适量,振摇使完全溶解,加水至刻度,摇匀,作为琥珀酸贮备液;取加有水20ml的100ml量瓶,称重,精密加入冰乙酸2ml,再称重,用水稀释至刻度,摇匀,精密量取6ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为乙酸贮备溶液;精密量取乙酸贮备液和琥珀酸贮备液各4.0ml,置同一25ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。照高效液相色谱法(中国药典2015年版四部通则0512)试验。以十八烷基硅烷键合硅胶为填充剂,以0.02moI/L磷酸二氢钾溶液(用6mol/L磷酸溶液调pH值至2.8)为流动相,流速每分钟1ml,检测波长为215nm。取对照溶液10μl, 注入液相色谱仪,按琥珀酸峰计算,理论板数不得少于8000。取供试品溶液与对照溶液各10μl,注入液相色谱仪,按干燥品计算,游离乙酸和琥珀酸总量不得过1.0%。[/color][color=#444444]计算公式: 游离乙酸含量=0.0768(WA/(W(1-干燥失重)))(γUA/γSA)[/color][color=#444444] 式中 WA为乙酸贮备溶液中冰乙酸量,mg;[/color][color=#444444] W为供试品的取样量,mg;[/color][color=#444444] γUA、γSA为供试品溶液、对照溶液中乙酸的峰面积。[/color][color=#444444] 游离琥珀酸含量=1.28(WS/(WUS(1-干燥失重)))(γUS/γSS)[/color][color=#444444] 式中 WS为琥珀酸贮备液中琥珀酸量,mg;[/color][color=#444444] WUS为供试品取样量,mg;[/color][color=#444444] γUS、γSS为供试品溶液、对照溶液琥珀酸的峰面积。[/color][color=#444444]我的问题是,根据“干燥品计算,游离乙酸和琥珀酸总量不得过1.0%”这句话,游离乙酸含量的最后计算的结果要不要乘以100%,比如我最后计算结果是0.0139,如果这个结果再乘以100%,就变为1.39%,从而超过限度,那么就需要重新做实验复核一遍。[/color]
测定水质中的硫化物,所配的试剂中,有一个是对氨基二甲基苯胺,买的是国外的试剂,用过一段时间后发现里面全都变的如黑沥青似的黏黏的,不知道是保存的哪个环节出了问题?(我们用的是黑色塑料瓶子,拧的很严,通风,干燥.)[em0810]
本人在8月发表的一篇原创中提及”甘氨酸与组氨酸无法分离“的问题,在经过10多天的准备,已有不小的收获,现在分享。摘要 目的: 建立用高效液相色谱法测定人凝血因子VIII中氨基酸含量。方法: 采用6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 为衍生剂,与氨基酸柱前衍生后,用Agilent 1200 高效液相色谱仪,AccQ·Tag C18柱( waters 150 mm ×3. 9 mm,4 μm) ,以水Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液和乙腈进行梯度洗脱,检测波长为248 nm,柱温37 ℃,进样量10μL。结果: 各氨基酸在32 min 内测定完毕,回收率为98.7% ~ 101.5%。RSD 均小于1. 5%。结论: 本法分离度好,快速、简便,可作为产品的质量控制方法。关键词: 6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯; 人凝血因子VIII; 甘氨酸; 衍生物; 梯度洗脱; 高效液相色谱法;氨基酸; 含量测定人凝血因子VIII,本品对缺乏人凝血因子礓所致的凝血机能障碍具有纠正作用,主要用于防治甲型血友病和获得性凝血因子Ⅷ缺乏而致的出血症状及这类病人的手术出血治疗。该药物制备过程中使用了氨基酸( 精氨酸、丙氨酸、甘氨酸、组氨酸、盐酸赖氨酸、脯氨酸 等) 做稳定剂,为了保证药品质量和用药安全,应对其中氨基酸的含量进行控制。该法依据过量的6 - 氨基喹啉基- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 在一定条件和氨基酸形成稳定的衍生产物( 柱前衍生) ,用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人凝血因子中各氨基酸的含量。1 仪器和试药1200 高效液相色谱系统( 美国Agilent 公司) ,配置低压四元梯度泵、1314B 紫外吸收检测器、自动进样器、柱温箱、Chemistations 化学工作站; Sartorius CP225D 电子微量天平( 德国Sartorius 公司) ; SartoriusPB - 21 型pH 计( 德国Sartorius 公司) ; LDZ5 -2 低速自动平衡离心机( 上海医用离心机厂) 等。各标准品均来自于中国食品药品检定研究院2 色谱条件及系统适用性试验色谱柱: Waters AccQ·Tag C18色谱柱( 3. 9 mm ×150 mm) ; 流动相: 水为溶剂D,Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液( A) - 乙腈( B) - 水( D) ,柱温:37 ℃; 检测波长: 248 nm。精密量取对照品溶液与供试品溶液10 μL,分别注入液相色谱仪,记录色谱图32 min。3 溶液制备3. 1 Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液称取三水乙酸钠190. 4 g,加注射用水1000 mL,搅拌,溶解,用稀磷酸将pH 调至5. 2,加入乙二胺四乙酸二钠溶液( 称取乙二胺四乙酸二钠100 mg,加注射用水100 mL,摇匀使其溶解) 10 mL,加入叠氮化钠0. 1 g 及三乙胺23. 7 mL( 17. 2 g) ,用稀磷酸滴定至pH 4. 95,用0. 45 μm 的滤膜过滤,于4 ℃储存,备用( 此条件下可保存6 个月) 。量取该溶液100 mL,加注射用水稀释至1000 mL,混匀,即得Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液。3. 2 对照品储备液混合对照品储备液精密称取各氨基酸对照品适量,置同一100 mL量瓶中,以注射用水溶解并定容至刻度。制成含氨基酸含量均含5. 0 mg·mL - 1 的混合对照品溶液,即得。单个对照品储备液: 精密称取各含氨基酸的各对照品适量,分别置100mL 量瓶中,用注射用水溶解并定容至刻度。制成分别含各氨基酸的单个对照品溶液,即得。3. 3 供试品储备液3. 3. 1 加样回收率试验溶液精密称取各氨基酸各0. 3200,0. 4000,0. 4800 g 和辅料适量,加人凝血因子VIII原液7. 5 mL,肝素钠适量,用1. 0 mol·L - 1 盐酸调pH 至6. 9,加0. 01 mol·L - 1枸橼酸三钠溶液溶解并定容于20 mL。分别制备成16. 0, 20. 0, 24. 0 mg·mL - 1溶液。3. 3. 2 空白溶液 按公司处方,加入辅料的混合物,用注射用水制备各空白溶液3. 4 内标溶液精密称取α - 氨基丁酸( AABA)0. 4 g,加注射用水定容至100 mL。4 氨基酸衍生方法4. 1 精密量取供试品储备液、样品及对照品储备液各1. 0 mL,加1. 5%磺基水杨酸9. 0 mL,混匀静置2 h以上, 3000 r·min - 1离心10 min,留取上清液。4. 2 精密量取“4. 1”项下上清液1. 0 mL( 其中对照品储备液对应上清液分别精密量取0. 06, 0. 4,0. 8,1. 0, 1. 2, 1. 6 mL) ,分别置10 mL 量瓶中,用注射用水定容。制备成供试品溶液、样品溶液及浓度分别为1. 5, 10. 0, 20. 0, 25. 0, 30. 0,40. 0 mg·mL - 1 的对照品溶液。4. 3 精密量取“4. 2”项下溶液各100 μL,分别加注射用水0. 4 mL 及内标溶液20 μL,混匀备用。4. 4 精密量取“4. 3”项下溶液30 μL 放入衍生管中,加硼酸缓冲液( pH 8 ~ 10) 210 μL 涡旋混合,并加入AQC 衍生剂60 μL 涡旋混合15 s,即为各供试品溶液,待用。







 我要推广仪器
我要推广仪器
 下载APP
下载APP